Welcome to SlamDunk docs.
SlamDunk also provides this documentation as package content for your convenience.The documentation can also be compiled into various formats using the Sphinx documentation generator .
SLAMseq is a novel sequencing protocol that directly uncovers 4-thiouridine incorporation events in RNA by high-throughput sequencing. When combined with metabolic labeling protocols, SLAM-seq allows to study the intracellular RNA dynamics, from transcription, RNA processing to RNA stability.
Original publication: Herzog et al., Nature Methods, 2017; doi:10.1038/nmeth.4435
Neumann, T., Herzog, V. A., Muhar, M., Haeseler, von, A., Zuber, J., Ameres, S. L., & Rescheneder, P. (2019). Quantification of experimentally induced nucleotide conversions in high-throughput sequencing datasets . BMC Bioinformatics, 20(1), 258. doi:10.1186/s12859-019-2849-7
This section covers the installation of slamdunk . Alternatively one could also run slamdunk from out of the box Docker containers.
There are 3 different possibilities:
We recommend using virtual environments to manage your Python installation. Our favourite is Anaconda , a cross-platform tool to manage Python environments. You can installation instructions for Anaconda here . Then you can directly install slamdunk from the Bioconda channel .
pip is the recommended tool for installing Python packages. It allows a convenient and simple installation of slamdunk from the Python Package Index PyPI .
To analyze a given SLAMSeq dataset with slamdunk using the default parameters which worked well on datasets during development, call the all dunk using the recommended minimum set of default parameters:
slamdunk all -r <reference fasta> -b <bed file> -o <output directory> -5 12 -n 100
-t <threads> -m -rl <maximum read length> --skip-sam files [files ...]
The input data you need and parameters you need to set yourself are the following:
| Parameter | Description |
|---|---|
| -r | The reference fasta file. |
| -b | BED-file containing coordinates for 3’ UTRs. |
| -o | The output directory where all output files of this dunk will be placed. |
| -t | The number of threads to use for this dunk. NextGenMap runs multi-threaded, so it is recommended to use more threads than available samples (default: 1). |
| files | Samplesheet (see Sample file format ) or a list of all sample BAM/FASTA(gz)/FASTQ(gz) files (wildcard * accepted). |
This will run the entire slamdunk analysis with the most relevant output files being:
If you want to have a very quick sanity check whether your desired conversion rates have been achieved, use this alleyoop command to plot the UTR conversion rates in your sample:
alleyoop utrrates -o <output directory> -r <reference fasta> -t <threads>
-b <bed file> -l <maximum read length> bam [bam ...]
| Parameter | Description |
|---|---|
| -o | The output directory where the plots will be placed. |
| -r | The reference fasta file. |
| -t | The number of threads to use. All tools run single-threaded, so it is recommended to use as many threads as available samples. |
| -b | BED-file containing coordinates for 3’ UTRs. |
| -l | Maximum read length in all samples. |
| bam | BAM file(s) containing the final filtered reads from the filter folder (wildcard * accepted). |
This will produce a plot like the following example where you can clearly see that individual conversions for a given starting base are balanced and unbiased, except for the T->C conversions in this SLAMSeq sample with longer labelling times.
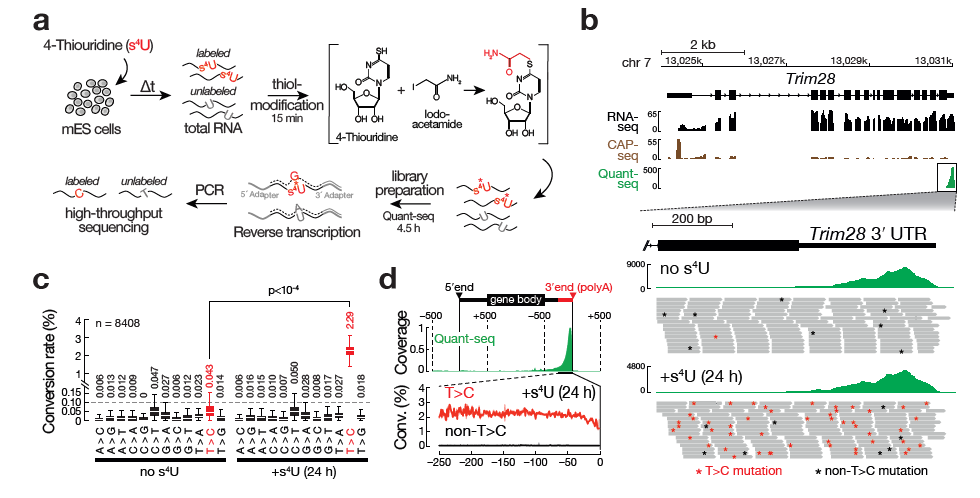
The individual measured conversion rates within a 3’ UTR are highly dependent on the T-content of a given UTR and the number of reads that cover those Ts. The more reads containing Ts, the more accurate the overall estimation of the conversion rate of T>Cs in this given 3’ UTR will be.
We have adressed this by using a 3’ UTR positions based view instead of a read based view: For each T-position in a given 3’ UTR, we record the number of reads covering this position and the number of reads with T>C conversion at this position - the more reads, the more accurate the T>C conversion rate estimation will be.
Both values are summed over all T-positions in the UTR - this way, less reliable T-positions with little reads will have lower impact on the overall T>C conversion rate estimation than T-positions with many reads.
Note: Since QuantSeq is a strand-specific assay, only sense reads will be considered for the final analysis!
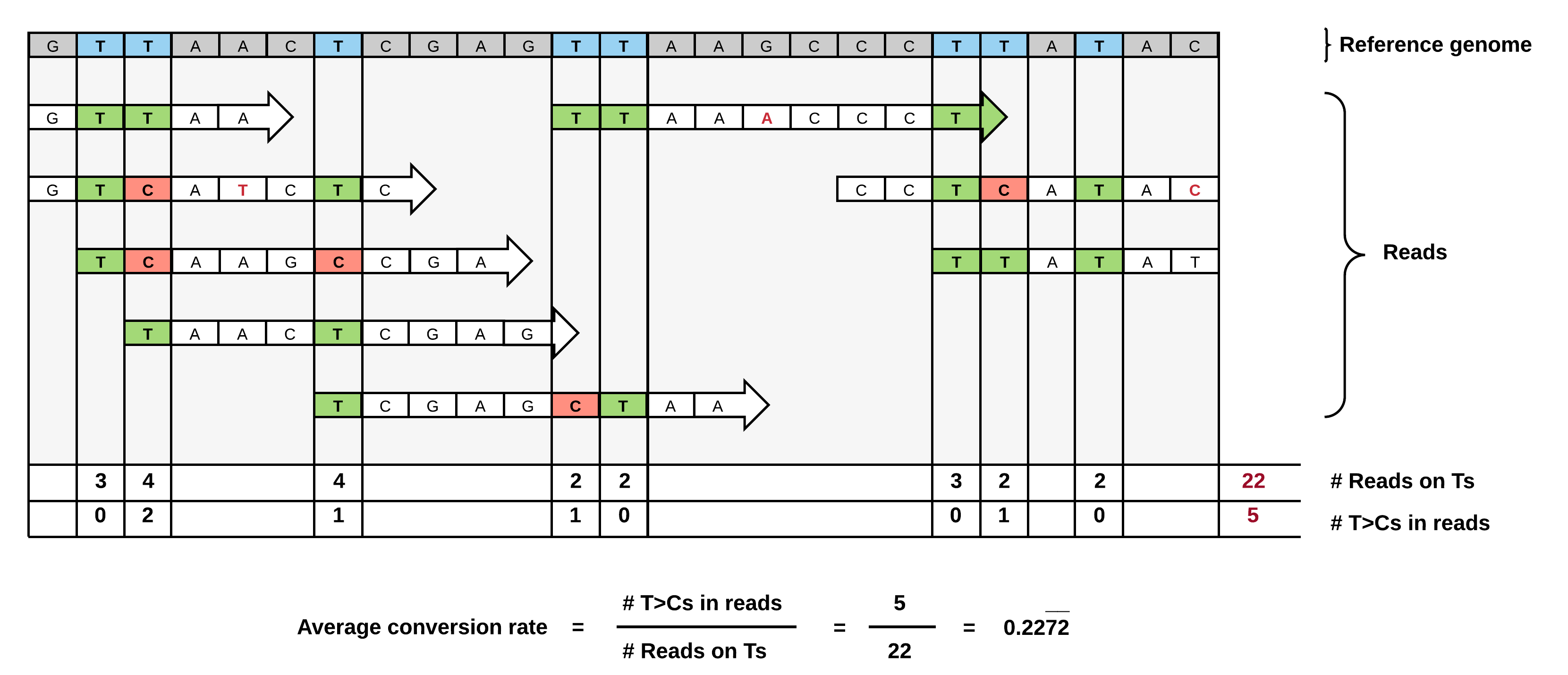
Multimappers are reads which align to more than one position in the genome with equal best alignment scores.
Such reads make up a substantial part of the alignment and are generally ignored, but can provide valuable information if they can be unequivocally reassigned to a given location, enhancing the overall signal and thus heavily influencing the overall accuracy.
However, there are also several caveats: For genes with isoforms or homologous genes, simply taking all alignments into account will heavily distort the signal, since the reads will likely not account equally for all regions, but rather originate mainly from a single region which cannot be unambiguously assigned.
We have settled for a very conservative multimapper reassignment strategy:
Since the QuantSeq technology specifically enriches for 3’ UTRs, we only consider alignments to annotated 3’ UTRs supplied to slamdunk as relevant.
Therefore, any multimappers with alignments to a single 3’ UTR and non-3’UTR regions (i.e. not annotated in the supplied reference) will be unequivocally assigned to the single 3’UTR. If there are multiple alignments to this single 3’UTR, one will be chosen at random.
For all other cases, were a read maps to several 3’ UTRs, we are unable to reassign the read uniquely to a given 3’UTR and thus discard it from the analysis.
In short, the procedure is as follows, illustrated by the example below:
Sample description files are a convenient way to provide multiple sample files as well as corresponding sample information to slamdunk . A sample description file is a tab or comma separated plain text file with the following columns:
| Column | Datatype | Description |
|---|---|---|
| Filepath | String | Path to raw reads in BAM/fasta(gz)/fastq(gz) format |
| Sample description | String | Description of the sample |
| Sample type | String | Type of sample. Only relevant for timecourses (pulse/chase) otherwise use any value |
| Timepoint | Integer | Timepoint of the sample in minutes. Again only relevant for timecourses otherwise use any value |
The tcount file is the central output file of slamdunk . It contains all results, conversion rates and other statistics for each UTR which is the main starting point for any subsequent analysis that will follow e.g. transcript half-life estimates or DE analysis.
Tcount files are essentially tab-separated text files containing one line entry per 3’ UTR supplied by the user. Each entry contains the following columns:
| Column | Datatype | Description |
|---|---|---|
| chromosome | String | Chromosome on which the 3’ UTR resides |
| start | Integer | Start position of the 3’ UTR |
| end | Integer | End position of the 3’ UTR |
| name | String | Name or ID of the 3’ UTR supplied by the user |
| length | Integer | Length of the 3’ UTR |
| strand | String | Strand of the 3’ UTR |
| conversionRate | Float | Average conversion rate (see Conversion rates ) |
| readsCPM | Float | Normalized number of reads as counts per million |
| Tcontent | Integer | Number of Ts in the 3’ UTR (As for - strand UTRs) |
| coverageOnTs | Integer | Cumulative coverage on all Ts in the 3’ UTR |
| conversionsOnTs | Integer | Cumulative number of T>C conversions in the 3’ UTR |
| readCount | Integer | Number of reads aligning to the 3’ UTR |
| tcReadCount | Integer | Number of reads with T>C conversions aligning to the 3’ UTR |
| multimapCount | Integer | Number of reads considered as multimappers aligning to the 3’ UTR |
Here is an example:
chr13 14197734 14199362 ENSMUSG00000039219 1628 + 0.0466045272969 4.00770947448 587 751 35 59 29 0
chr2 53217389 53219220 ENSMUSG00000026960 1831 + 0.0270802560315 28.3936027175 709 6093 165 418 138 2
chr2 53217389 53218446 ENSMUSG00000026960 1057 + 0.0268910814471 23.9783295677 407 5169 139 353 118 0
chr3 95495394 95495567 ENSMUSG00000015522 173 + 0.0290697674419 1.08683646766 53 172 5 16 5 0
chr3 95495394 95497237 ENSMUSG00000015522 1843 + 0.0253636702723 11.6834920273 584 2681 68 172 55 4
chr6 113388777 113389056 ENSMUSG00000079426 279 + 0.0168514412417 16.7780379694 71 2255 38 247 38 3
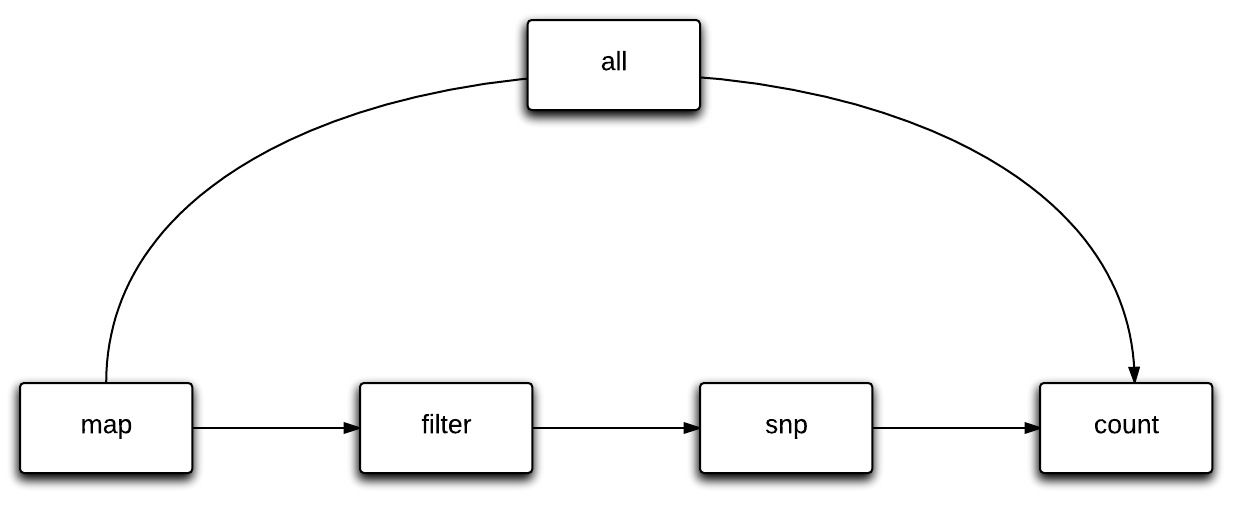
Slamdunk is a modular analysis software. Modules are dubbed dunks and each dunk builds upon the results from the previous dunks.
The idea is to always process all samples with one command line call of a given dunk. Output files typically get the same name as the input files with a certain prefix (e.g. “slamdunk_mapped”). The command line interface follows the “samtools/bwa” style. Meaning that all commands are available through the central executable/script slamdunk (located in the bin directory) Wildcard characters to supply multiple files are recognized throughout slamdunk .
Available dunks are:
'map', 'filter', 'snp', 'count', 'all'
Calling a module with –help shows all possible parameters text:
usage: slamdunk all [-h] -r REFERENCEFILE -b BED [-fb FILTERBED] -o OUTPUTDIR [-5 TRIM5]
[-a MAXPOLYA] [-n TOPN] [-t THREADS] [-q] [-e] [-m]
[-mq MQ] [-mi IDENTITY] [-nm NM] [-mc COV] [-mv VAR]
[-mts] [-rl MAXLENGTH] [-mbq MINQUAL] [-i SAMPLEINDEX]
[-ss]
files [files ...]
positional arguments:
files Single csv/tsv file (recommended) containing all
sample files and sample info or a list of all sample
BAM/FASTA(gz)/FASTQ(gz) files
optional arguments:
-h, --help show this help message and exit
-r REFERENCEFILE, --reference REFERENCEFILE
Reference fasta file
-b BED, --bed BED BED file with 3'UTR coordinates
-fb FILTERBED, --filterbed FILTERBED
BED file with 3'UTR coordinates to filter multimappers
-o OUTPUTDIR, --outputDir OUTPUTDIR
Output directory for slamdunk run.
-5 TRIM5, --trim-5p TRIM5
Number of bp removed from 5' end of all reads
(default: 12)
-a MAXPOLYA, --max-polya MAXPOLYA
Max number of As at the 3' end of a read (default: 4)
-n TOPN, --topn TOPN Max. number of alignments to report per read (default:
1)
-t THREADS, --threads THREADS
Thread number (default: 1)
-q, --quantseq Run plain Quantseq alignment without SLAM-seq scoring
-e, --endtoend Use a end to end alignment algorithm for mapping.
-m, --multimap Use reference to resolve multimappers (requires -n >
1).
-mq MQ, --min-mq MQ Minimum mapping quality (default: 2)
-mi IDENTITY, --min-identity IDENTITY
Minimum alignment identity (default: 0.95)
-nm NM, --max-nm NM Maximum NM for alignments (default: -1)
-mc COV, --min-coverage COV
Minimimum coverage to call variant (default: 10)
-mv VAR, --var-fraction VAR
Minimimum variant fraction to call variant (default:
0.8)
-mts, --multiTCStringency
Multiple T>C conversion required for T>C read
-rl MAXLENGTH, --max-read-length MAXLENGTH
Max read length in BAM file
-mbq MINQUAL, --min-base-qual MINQUAL
Min base quality for T -> C conversions (default: 27)
-i SAMPLEINDEX, --sample-index SAMPLEINDEX
Run analysis only for sample <i>. Use for distributing
slamdunk analysis on a cluster (index is 1-based).
-ss, --skip-sam Output BAM while mapping. Slower but, uses less hard
disk.
The flow of slamdunk is to first map your reads, filter your alignments, call variants on your final alignments and use these to calculate conversion rates, counts and various statistics for your 3’UTRs.
All steps create a log file that has the same name as the output file. Typically there is one log file per sample and task (makes parallel execution easier). Command line output is limited to a minimum at the moment. If a sample is finished a ”.” is printed (very basic progress bar).
The map dunk is used to map reads to a given genome using NextGenMap’s SLAMSeq alignment settings.
slamdunk map [-h] -r <reference fasta> -o <output directory> [-5 <bp to trim from 5' end>]
[-n <Output up to N alignments per multimapper>] [-a <maximum number of 3' As before trimming] [-t <threads>]
[-q] [-e] [-i <sample index>] [-ss] files [files ...]
| File | Description |
|---|---|
| Reference fasta | The reference sequence of the genome to map against in fasta format. |
| files | Samplesheet (see Sample file format ) or a list of all sample BAM/FASTA(gz)/FASTQ(gz) files (wildcard * accepted). |
| File | Description |
|---|---|
| Mapped BAM file | One BAM file per sample containing the mapped reads. |
| Mapped bam index file | The raw unmapped reads in fastq / BAM format (multiple read files can be run at once). |
Output files have the same name as the input files with the prefix “_slamdunk_mapped.bam”. For example:
34330_An312_wt-2n_mRNA-slamseq-autoquant_0h-R1.fq.gz ->
34330_An312_wt-2n_mRNA-slamseq-autoquant_0h-R1.fq_slamdunk_mapped.bam
| Parameter | Required | Description |
|---|---|---|
| -h | Prints the help. | |
| -r | x | The reference fasta file. |
| -o | x | The output directory where all output files of this dunk will be placed. |
| -5 | Number of bases that will be hard-clipped from the 5’ end of each read. | |
| -a | Maximum number of As at the 3’ end of a read. | |
| -n | The maximum number of alignments that will be reported for a multi-mapping read (i.e. reads with multiple alignments of equal best scores). | |
| -t | The number of threads to use for this dunk. NextGenMap runs multi-threaded, so it is recommended to use more threads than available samples. | |
| -q | Deactivates NextGenMap’s SLAMSeq alignment settings. Can be used to align plain QuantSeq data instead of SLAMSeq data. | |
| -e | Switches to semi-global alignment instead of local alignment. | |
| -i | Run analysis only for sample <i>. Use for distributin slamdunk analysis on a cluster (index is 0-based). | |
| -ss | Output BAM while mapping. Slower, but uses less hard disk. | |
| file | x | Samplesheet (see Sample file format ) or a list of all sample BAM/FASTA(gz)/FASTQ(gz) files (wildcard * accepted). |
The filter dunk is used to filter the raw alignments from the map dunk using multiple quality criteria to obtain the final alignments for all subsequent analyses.
slamdunk filter [-h] -o <output directory> [-b <bed file>] [-mq <MQ cutoff>] [-mi <identity cutoff>]
[-nm <NM cutoff>] [-t <threads>] bam [bam ...]
| File | Description |
|---|---|
| bam | The raw mapped reads in BAM format from the map dunk. |
| -b | (optional) Bed file with coordinates of 3’UTRs. |
| File | Description |
|---|---|
| Filtered BAM file | One BAM file per sample containing the filtered reads. |
| Filtered bam index file | One BAM index file accompanying each filtered BAM file. |
Output files have the same name as the input files with the prefix “_filtered”. For example:
34330_An312_wt-2n_mRNA-slamseq-autoquant_0h-R1.fq_slamdunk_mapped.bam ->
34330_An312_wt-2n_mRNA-slamseq-autoquant_0h-R1.fq_slamdunk_mapped_filtered.bam
| Parameter | Required | Description |
|---|---|---|
| -h | Prints the help. | |
| -o | x | The output directory where all output files of this dunk will be placed. |
| -b | BED-file containing coordinates for 3’ UTRs. | |
| -mq | Minimum mapping quality required to retain a read. | |
| -mi | Minimum alignment identity required to retain a read. | |
| -nm | Maximum number of mismatches allowed in a read. | |
| -t | The number of threads to use for this dunk. This dunk runs single-threaded so the number of threads should be equal to the number of available samples. | |
| bam | x | BAM file(s) containing the raw mapped reads (wildcard * accepted). |
The snp dunk is used to call variants on the final filtered alignments of the filter dunk using VarScan2 . Any called T->C SNPs from this dunk will be excluded in the subsequent analyses to reduce the false-positive number.
slamdunk snp [-h] -o <output directory> -r <reference fasta> [-c <coverage cutoff>]
[-f <variant fraction cutoff>] [-t <threads>] bam [bam ...]
| File | Description |
|---|---|
| bam | The final filtered reads in BAM format from the filter dunk. |
| File | Description |
|---|---|
| VCF file | One VCF file per sample containing the called variants. |
Output files have the same name as the input files with the prefix “_snp”. For example:
34330_An312_wt-2n_mRNA-slamseq-autoquant_0h-R1.fq_slamdunk_mapped_filtered.bam ->
34330_An312_wt-2n_mRNA-slamseq-autoquant_0h-R1.fq_slamdunk_mapped_filtered_snp.vcf
| Parameter | Required | Description |
|---|---|---|
| -h | Prints the help. | |
| -r | x | The reference fasta file. |
| -o | x | The output directory where all output files of this dunk will be placed. |
| -c | Minimum coverage to call a variant. | |
| -f | Minimum variant fraction to call a variant. | |
| -t | The number of threads to use for this dunk. VarScan2 runs multi-threaded, so it is recommended to use more threads than available samples. | |
| bam | x | BAM file(s) containing the final filtered reads (wildcard * accepted). |
The count dunk calculates all relevant numbers on statistics of SLAMSeq reads for each given 3’ UTR. Central output will be tcount table (see Tcount file format ).
slamdunk count [-h] -o <output directory> [-s <SNP directory>] -r <reference fasta> -b <bed file> [-m]
[-l <maximum read length>] [-q <minimum base quality>] [-t <threads] bam [bam ...]
Note: Since QuantSeq is a strand-specific assay, only sense reads will be considered for the final analysis!
| File | Description |
|---|---|
| bam | The final filtered reads in BAM format from the filter dunk. |
| -s | (optional) The called variants from the snp dunk to filter false-positive T->C conversions. |
| -b | Bed file with coordinates of 3’UTRs. |
| File | Description |
|---|---|
| Tcount file | A tab-separated tcount file per sample containing the SLAMSeq statistics (see Tcount file format ). |
| Bedgraph file | A bedgraph file per sample showing the T->C conversion rate on each covered reference T nucleotide. |
Output files have the same name as the input files with the prefix “_tcount”. For example:
34330_An312_wt-2n_mRNA-slamseq-autoquant_0h-R1.fq_slamdunk_mapped_filtered.bam ->
34330_An312_wt-2n_mRNA-slamseq-autoquant_0h-R1.fq_slamdunk_mapped_filtered_tcount.csv
| Parameter | Required | Description |
|---|---|---|
| -h | Prints the help. | |
| -o | x | The output directory where all output files of this dunk will be placed. |
| -s | The output directory of the snp dunk containing the called variants. | |
| -r | x | The reference fasta file. |
| -b | x | BED-file containing coordinates for 3’ UTRs. |
| -l | Maximum read length (will be automatically estimated if not set). | |
| -m | Flag to activate the multiple T->C conversion stringency: Only T->C conversions in reads with more than 1 T->C conversion will be counted. | |
| -q | Minimum base quality for T->C conversions to be counted. | |
| -t | The number of threads to use for this dunk. This dunk runs single-threaded so the number of threads should be equal to the number of available samples. | |
| bam | x | BAM file(s) containing the final filtered reads (wildcard * accepted). |
The all dunk is used to run an entire slamdunk run at once. It sequentially calls the map , filter , snp and count dunks and provides parameters to keep full control over all dunks.
slamdunk all [-h] -r <reference fasta> -b <bed file> [-fb <bed file>] -o <output directory> [-5 <bp to trim from 5' end>] [-a <maximum number of 3' As before trimming]
[-n <Output up to N alignments per multimapper>] [-t <threads>] [-q] [-e] [-m] [-mq <MQ cutoff>]
[-mi <identity cutoff>] [-nm <NM cutoff>] [-mc <coverage cutoff>] [-mv <variant fraction cutoff>] [-mts]
[-rl <maximum read length>] [-mbq <minimum base quality>] [-i <sample index>] [-ss] files [files ...]
| File | Description |
|---|---|
| Reference fasta | The reference sequence of the genome to map against in fasta format. |
| -b | Bed file with coordinates of 3’UTRs. |
| file | Samplesheet (see Sample file format ) or a list of all sample BAM/FASTA(gz)/FASTQ(gz) files (wildcard * accepted). |
One separate directory will be created for each dunk output:
| Folder | Dunk |
|---|---|
| map | map |
| filter | filter |
| snp | snp |
| count | count |
| Parameter | Required | Description |
|---|---|---|
| -h | x | Prints the help. |
| -r | x | The reference fasta file. |
| -b | x | BED-file containing coordinates for 3’ UTRs. |
| -fb | BED-file used to filter multimappers. | |
| -o | x | The output directory where all output files of this dunk will be placed. |
| -5 | Number of bases that will be hard-clipped from the 5’ end of each read [map] . | |
| -a | Maximum number of A at the 3’ end of a read. | |
| -n | The maximum number of alignments that will be reported for a multi-mapping read (i.e. reads with multiple alignments of equal best scores) [map] . | |
| -t | The number of threads to use for this dunk. NextGenMap runs multi-threaded, so it is recommended to use more threads than available samples | |
| -q | Deactivates NextGenMap’s SLAMSeq alignment settings. Can be used to align plain QuantSeq data instead of SLAMSeq data [map] . | |
| -e | Switches to semi-global alignment instead of local alignment [map] . | |
| -m | Use 3’UTR annotation to filter multimappers [filter] . | |
| -mq | Minimum mapping quality required to retain a read [filter] . | |
| -mi | Minimum alignment identity required to retain a read [filter] . | |
| -nm | Maximum number of mismatches allowed in a read [filter] . | |
| -mc | Minimum coverage to call a variant [snp] . | |
| -mv | Minimum variant fraction to call a variant [snp] . | |
| -mts | Flag to activate the multiple T->C conversion stringency: Only T->C conversions in reads with more than 1 T->C conversion will be counted. [count] . | |
| -rl | Maximum read length (will be automatically estimated if not set) [count] . | |
| -mbq | Minimum base quality for T->C conversions to be counted [count] . | |
| files | x | Samplesheet (see Sample file format ) or a list of all sample BAM/FASTA(gz)/FASTQ(gz) files (wildcard * accepted). |
Alleyoop ( A dditional s L amdunk he L p E r tools for an Y diagn O stics O r P lots) is a collection of tools for post-processing and running diagnostics on slamdunk analyses. Similar to slamdunk , the command line interface follows the “samtools/bwa” style. Meaning that all commands are available through the central executable/script alleyoop (located in the bin directory).
MultiQC:
We implemented a module for MultiQC to support summary reports of Alleyoop modules. Currently MultiQC supports the summary , rates , utrrates , tcperreadpos and tcperutrpos commands.
This tool allows you to deduplicate a given bam-file. While many tools like Picard tools already collapses reads with same start and end position on the chromosome, alleyoop only collapses reads with same start and end position, mapping to the same strand and identical read sequence.
alleyoop dedup [-h] -o <output directory> [-tc <# tc mutations>] [-t <threads>] bam [bam ...]
| File | Description |
|---|---|
| bam | Fastq/BAM file(s) containing the final filtered reads from slamdunk . Can be multiple if multiple samples are analysed simultaneously (wildcard * is recognized). |
| File | Description |
|---|---|
| Deduplicated BAM file | One BAM file per sample containing the deduplicated reads. |
Output files have the same name as the input files with the prefix “_dedup”. For example:
34330_An312_wt-2n_mRNA-slamseq-autoquant_0h-R1.fq_slamdunk_mapped_filtered.bam ->
34330_An312_wt-2n_mRNA-slamseq-autoquant_0h-R1.fq_slamdunk_mapped_filtered_dedup.bam
| Parameter | Required | Description |
|---|---|---|
| -h | Prints the help. | |
| -o | x | The output directory where all output files of this tool will be placed. |
| -tc | Only select reads with -tc number of T>C mutations for deduplication. | |
| -t | The number of threads to use. All tools run single-threaded, so it is recommended to use as many threads as available samples. | |
| bam | x | Fastq/BAM file(s) containing the final filtered reads (wildcard * accepted). |
This tool allows you to collapse all 3’UTR entries of a tcount file into one single entry per 3’UTR (similar to the exons->gene relationship in a gtf-file). All entries with identical 3’UTR IDs will be merged.
alleyoop collapse [-h] -o <output directory> [-t <threads>] tcount [tcount ...]
| File | Description |
|---|---|
| Tcount file | A tab-separated tcount file per sample containing the SLAMseq statistics (see Tcount file format ). |
| File | Description |
|---|---|
| Tcount file | A tab-separated tcount file per sample containing the SLAMseq statistics (see Tcount file format ) with collapsed 3’UTR entries. |
Output files have the same name as the input files with the prefix “_collapsed”.
For example:
34330_An312_wt-2n_mRNA-slamseq-autoquant_0h-R1.fq_slamdunk_mapped_filtered_tcount.tsv ->
34330_An312_wt-2n_mRNA-slamseq-autoquant_0h-R1.fq_slamdunk_mapped_filtered_tcount_collapsed.tsv
| Parameter | Required | Description |
|---|---|---|
| -h | Prints the help. | |
| -o | x | The output directory where all output files of this tool will be placed. |
| -t | The number of threads to use. All tools run single-threaded, so it is recommended to use as many threads as available samples. | |
| Tcount file | x | A tab-separated tcount file per sample containing the SLAMseq statistics (see Tcount file format ) with collapsed 3’UTR entries. |
This tool computes the overall conversion rates in your reads and plots them as a barplot.
alleyoop rates [-h] -o <output directory> -r <reference fasta> [-mq <MQ cutoff>]
[-t <threads>] bam [bam ...]
| File | Description |
|---|---|
| Reference fasta | The reference sequence of the genome to map against in fasta format. |
| bam | Fastq/BAM file(s) containing the final filtered reads from slamdunk (wildcard * accepted). |
| File | Description |
|---|---|
| overallrates.csv | Tab-separated table of the overall conversion rates. |
| overallrates.pdf | Overall conversion rate plot file. |
Below is an example plot of the overall conversion rates of the reads in a sample. One can appreciate the typical excess of T->C conversion (A->G on minus strand) of the SLAMseq technology for later labelling timepoints.
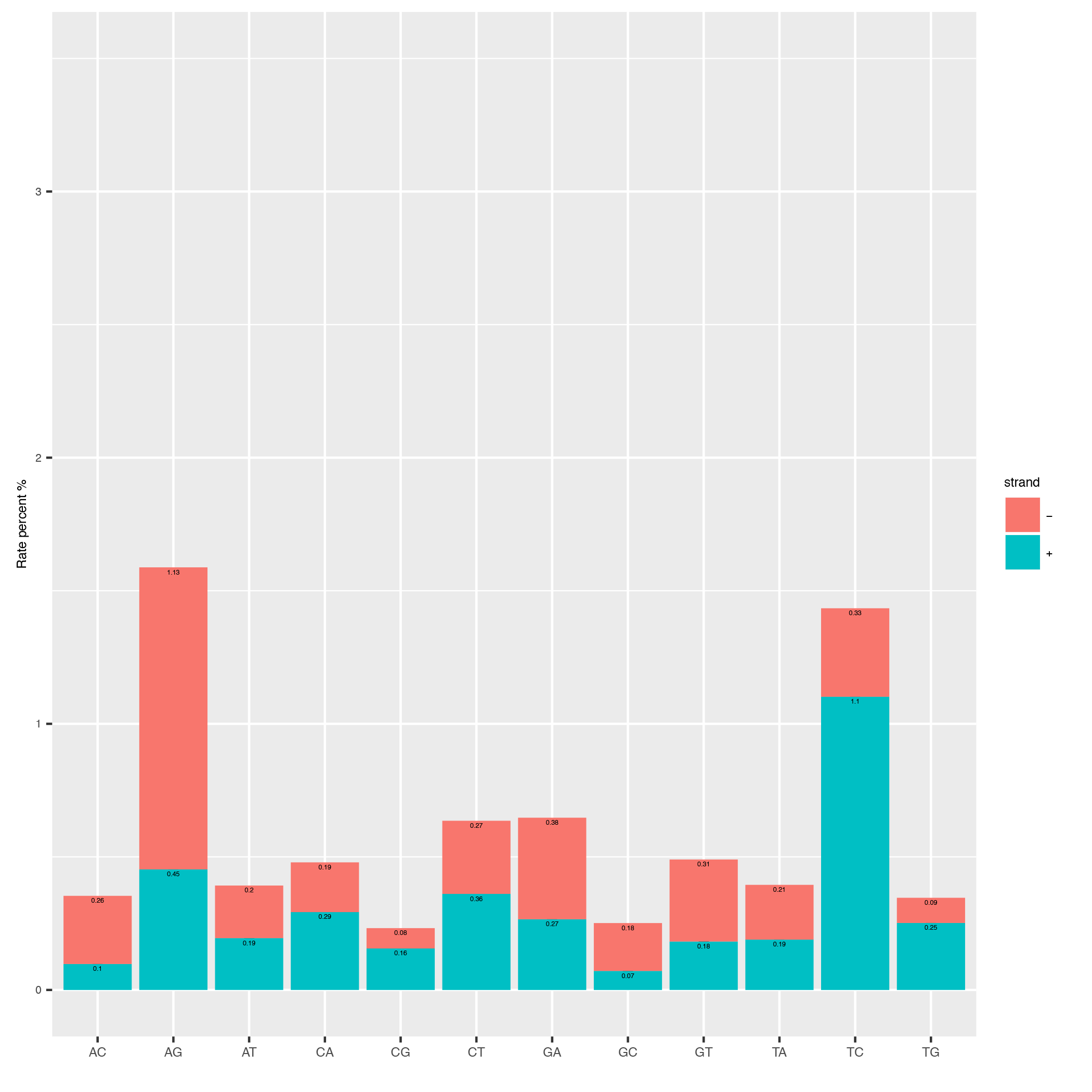
| Parameter | Required | Description |
|---|---|---|
| -h | Prints the help. | |
| -o | x | The output directory where all output files of this tool will be placed. |
| -r | x | The reference fasta file. |
| -mq | Minimum base quality for T->C conversions to be counted. | |
| -t | The number of threads to use. All tools run single-threaded, so it is recommended to use as many threads as available samples. | |
| bam | x | Fastq/BAM file(s) containing the final filtered reads. Can be multiple if multiple samples are analysed simultaneously (wildcard * is recognized). |
This tool computes the genomic context of all Ts in a read and plots them as barplot to inspect any biases in that direction.
alleyoop tccontext [-h] -o <output directory> -r <reference fasta> [-mq <MQ cutoff>]
[-t <threads>] bam [bam ...]
| File | Description |
|---|---|
| Reference fasta | The reference sequence of the genome to map against in fasta format. |
| bam | BAM file(s) containing the final filtered reads from slamdunk (wildcard * accepted). |
| File | Description |
|---|---|
| tccontext.csv | Tab-separated table of the 5’ and 3’ T-contexts, separated by strand. |
| tccontext.pdf | T-context plot file. |
Below is an example plot of the T-context of all reads in a sample. On top you will find the 5’ context of individual Ts, at the bottom the respective 3’ context of the individual Ts. Note, that these will not be reciprocal (see e.g. this publication ).
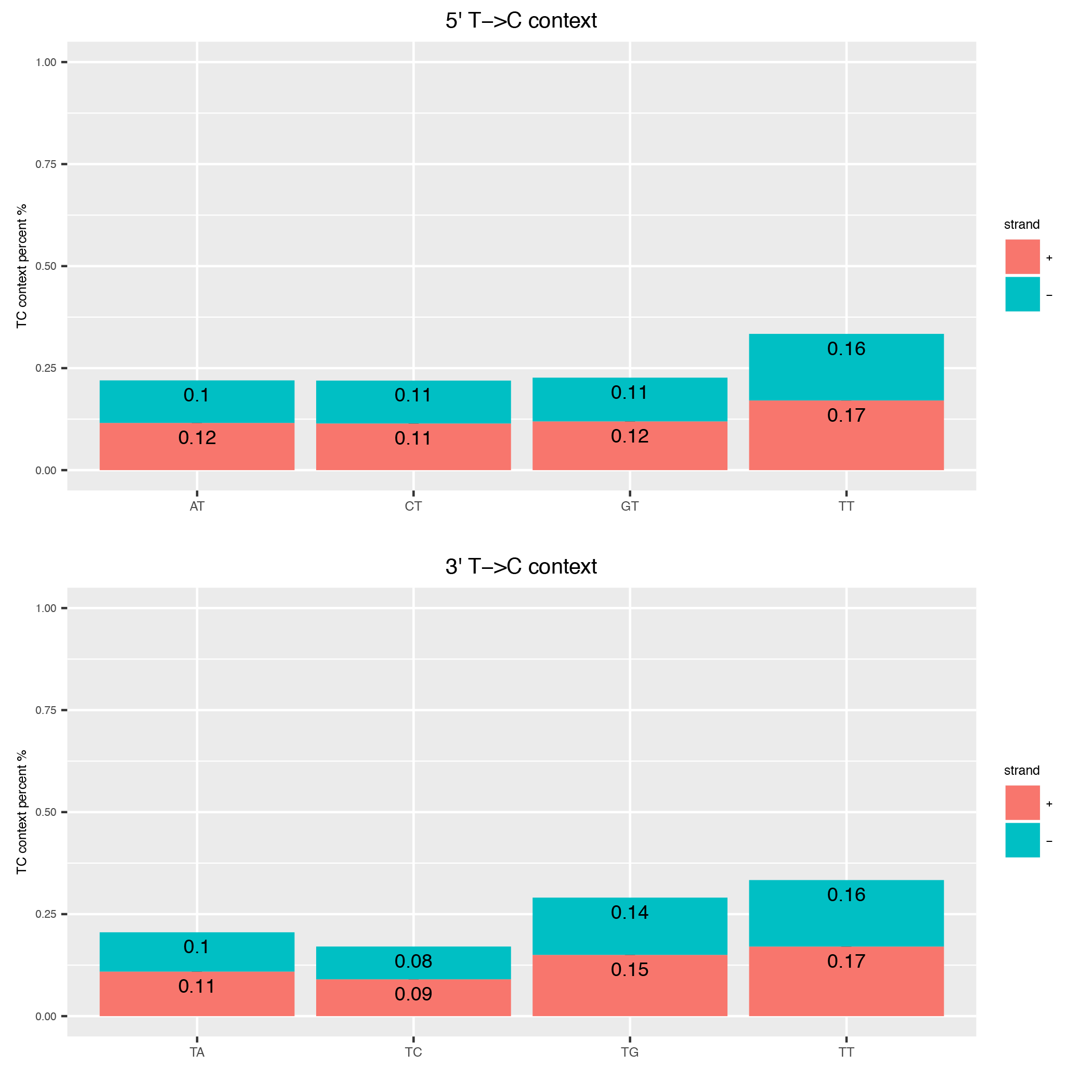
| Parameter | Required | Description |
|---|---|---|
| -h | Prints the help. | |
| -o | x | The output directory where all output files of this tool will be placed. |
| -r | x | The reference fasta file. |
| -mq | Minimum base quality for T->C conversions to be counted. | |
| -t | The number of threads to use. All tools run single-threaded, so it is recommended to use as many threads as available samples. | |
| bam | x | BAM file(s) containing the final filtered reads (wildcard * accepted). |
This tool checks the individual conversion rates per 3’UTR and plots them as boxplots over the entire realm of 3’UTRs. Each conversion is normalized to all possible conversions from it’s starting base e.g. A->G / (A->A + A->G + A->C + A->T).
alleyoop utrrates [-h] -o <output directory> [-r <reference fasta>] [-mq <MQ cutoff>] [-m]
[-t <threads>] -b <bed file> -l <maximum read length> bam [bam ...]
| File | Description |
|---|---|
| Reference fasta | The reference sequence of the genome to map against in fasta format. |
| -b | Bed file with coordinates of 3’UTRs. |
| bam | BAM file(s) containing the final filtered reads from slamdunk (wildcard * accepted). |
| File | Description |
|---|---|
| mutationrates_utr.csv | Tab-separated table with conversion reads, one UTR per line. |
| mutationrates_utr.pdf | UTR conversion rate plot file. |
Below is an example plot of conversion rates for all UTRs for a given sample. Typically, the individual conversions for a given starting base are balanced and unbiased, except for T->C conversions in SLAMseq samples with longer labelling times.
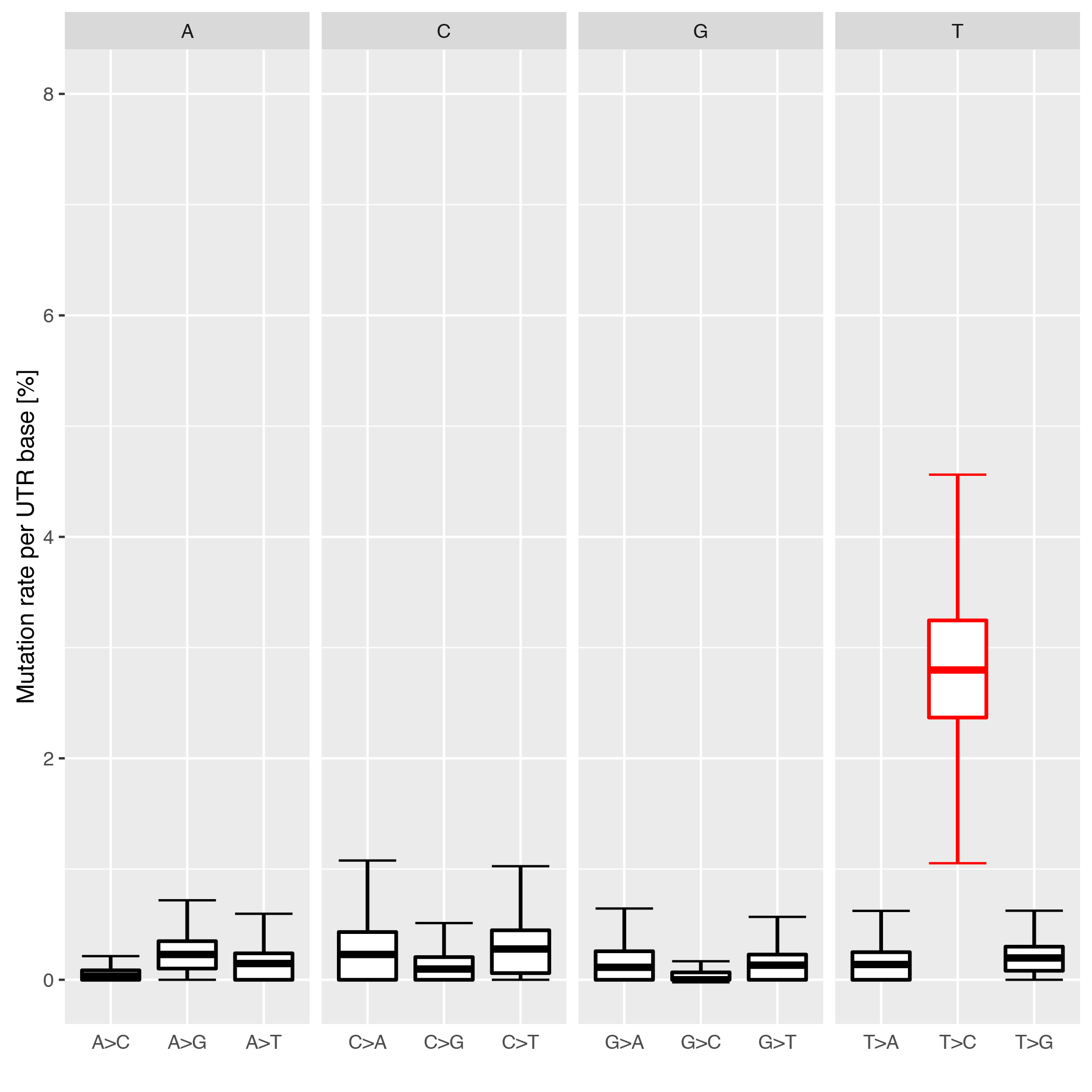
| Parameter | Required | Description |
|---|---|---|
| -h | Prints the help. | |
| -o | x | The output directory where all output files of this tool will be placed. |
| -r | x | The reference fasta file. |
| -mq | Minimum base quality for T->C conversions to be counted. | |
| -m | Flag to activate the multiple T->C conversion stringency: Only T->C conversions in reads with more than 1 T->C conversion will be counted. | |
| -t | The number of threads to use. All tools run single-threaded, so it is recommended to use as many threads as available samples. | |
| -b | x | Bed file with coordinates of 3’UTRs. |
| -l | Maximum read length in all samples (will be automatically estimated if not set). | |
| bam | x | BAM file(s) containing the final filtered reads (wildcard * accepted). |
This tool produces some QA about the quality of your variant calls: Ideally, your T>C SNP calls should be independently of the number of reads with T>C conversions found in an UTR. Otherwise, this would mean that you call more T>C SNPs the more T>C reads you have and thus you lose signal by falsely calling SNPs from true T>C conversions.
To assess this, the UTRs are ranked by the number of containing T>C reads and marked with a bar if also a T>C SNP was called in the respective UTR. The list is then filtered for 3’UTRs with sufficient coverage to confidently call SNPs by using only the upper quartile of the 3’UTRs according to total read coverage.
The resulting plots will show once the distribution of SNPs across ranked 3’UTRs being blind to SNP information and including SNP information. Ideally, one would see the SNPs biased towards 3’UTRs with high T>C read content in the blind situation and uniformly distributed across all 3’UTRs in the informed situation.
This difference is also quantified using a GSEA-like Mann-Whitney-U test.
alleyoop snpeval [-h] -o <output directory> -s <SNP directory> -r <reference fasta> -b <bed file> [-c <coverage cutoff>]
[-f <variant fraction cutoff>] [-m] [-l <maximum read length>] [-q <minimum base quality>] [-t <threads>]
bam [bam ...]
| File | Description |
|---|---|
| Reference fasta | The reference sequence of the genome to map against in fasta format. |
| -s | Directory of called SNPs from snp dunk. |
| -b | Bed file with coordinates of 3’UTRs. |
| bam | BAM file(s) containing the final filtered reads from slamdunk (wildcard * accepted). |
| File | Description |
|---|---|
| SNPeval.csv | Tab-separated table with read counts, T>C read counts and SNP indication, one UTR per line. |
| SNPeval.pdf | SNP evaluation plot. |
An example plot is coming soon!
| Parameter | Required | Description |
|---|---|---|
| -h | Prints the help. | |
| -o | x | The output directory where all output files of this tool will be placed. |
| -s | x | The output directory of the snp dunk containing the called variants. |
| -r | x | The reference fasta file. |
| -q | Minimum base quality for T->C conversions to be counted. | |
| -m | Flag to activate the multiple T->C conversion stringency: Only T->C conversions in reads with more than 1 T->C conversion will be counted. | |
| -c | Minimum coverage to call a variant. | |
| -f | Minimum variant fraction to call a variant. | |
| -t | The number of threads to use. All tools run single-threaded, so it is recommended to use as many threads as available samples. | |
| -b | x | Bed file with coordinates of 3’UTRs. |
| -l | Maximum read length in all samples (will be automatically estimated if not set). | |
| bam | x | BAM file(s) containing the final filtered reads (wildcard * accepted). |
This tool lists basic statistics of the mapping process in a text file.
alleyoop summary [-h] -o <output file> [-t <directory of tcount files>] bam [bam ...]
| File | Description |
|---|---|
| bam | BAM file(s) containing the final filtered reads from slamdunk (wildcard * accepted). |
| tcount file directory | (optional) Directory containing the associated tcount file(s) to the input BAM file(s). |
| File | Description |
|---|---|
| outputfile | Tab-separated table with mapping statistics. |
| outputfile_PCA.pdf | PCA plot of the samples based on T>C read counts per UTR. |
| outputfile_PCA.txt | PCA values of the samples based on T>C read counts per UTR. |
The output file will be a tab-separated text file with the following columns:
| Column | Content |
|---|---|
| FileName | Path to raw reads in BAM/fasta(gz)/fastq(gz) format. |
| SampleName | Description of the sample. |
| SampleType | Type of sample. |
| SampleTime | Timepoint of the sample in minutes. |
| Sequenced | Number of sequenced reads. |
| Mapped | Number of mapped reads. |
| Deduplicated | Number of deduplicated reads. |
| Filtered | Number of retained reads after filtering. |
| Counted | Number of counted reads within UTRs (optional: only if tcount file directory was supplied) . |
| Annotation | Annotation used for filtering. |
An example PCA plot is coming soon!
This tool merges tcount files of multiple samples into a single table based upon an expression of columns.
alleyoop merge [-h] -o <output file> [-c <expression>] countFiles [countFiles ...]
| File | Description |
|---|---|
| countFiles | A tab-separated tcount file per sample containing the SLAMseq statistics (see Tcount file format ). |
| File | Description |
|---|---|
| outputfile | Tab-separated table merged tcount information based upon expression. |
| Parameter | Required | Description |
|---|---|---|
| -h | Prints the help. | |
| -o | x | The output file name. |
| -c | Column or expression used to summarize files (e.g. “TcReadCount / ReadCount”) | |
| countFiles | x | A tab-separated tcount file per sample containing the SLAMseq statistics (see Tcount file format ). |
This tool calculates the individual mutation rates per position in a read treating T->C mutations separately. This plot can be used to search for biases along reads.
alleyoop tcperreadpos [-h] -r <reference fasta> [-s <SNP directory>]
[-l <maximum read length>] -o <output directory> [-mq <MQ cutoff>]
[-t <threads>] bam [bam ...]
| File | Description |
|---|---|
| Reference fasta | The reference sequence of the genome to map against in fasta format. |
| -s | (optional) The called variants from the snp dunk to filter false-positive T->C conversions. |
| bam | BAM file(s) containing the final filtered reads from slamdunk (wildcard * accepted). |
| File | Description |
|---|---|
| tcperreadpos.csv | Tab-separated table with mutation rates, one line per read position. |
| tcperreadpos.pdf | Plot of the mutation rates along the reads. |
Below is an example plot of mutation rates along all reads in a sample. Typically, one will see increasing error rates towards the end of a reads, as for all Illumina reads. In addition, depending on how many bases were clipped from the 5’ end of the reads, one will also observe higher error rates in the beginning of the read as illustrated in the example plot. Finally, for SLAMseq samples with longer labelling times, the overall T->C conversions in the bottom plot will begin to increase compared to the overall background in the top plot.
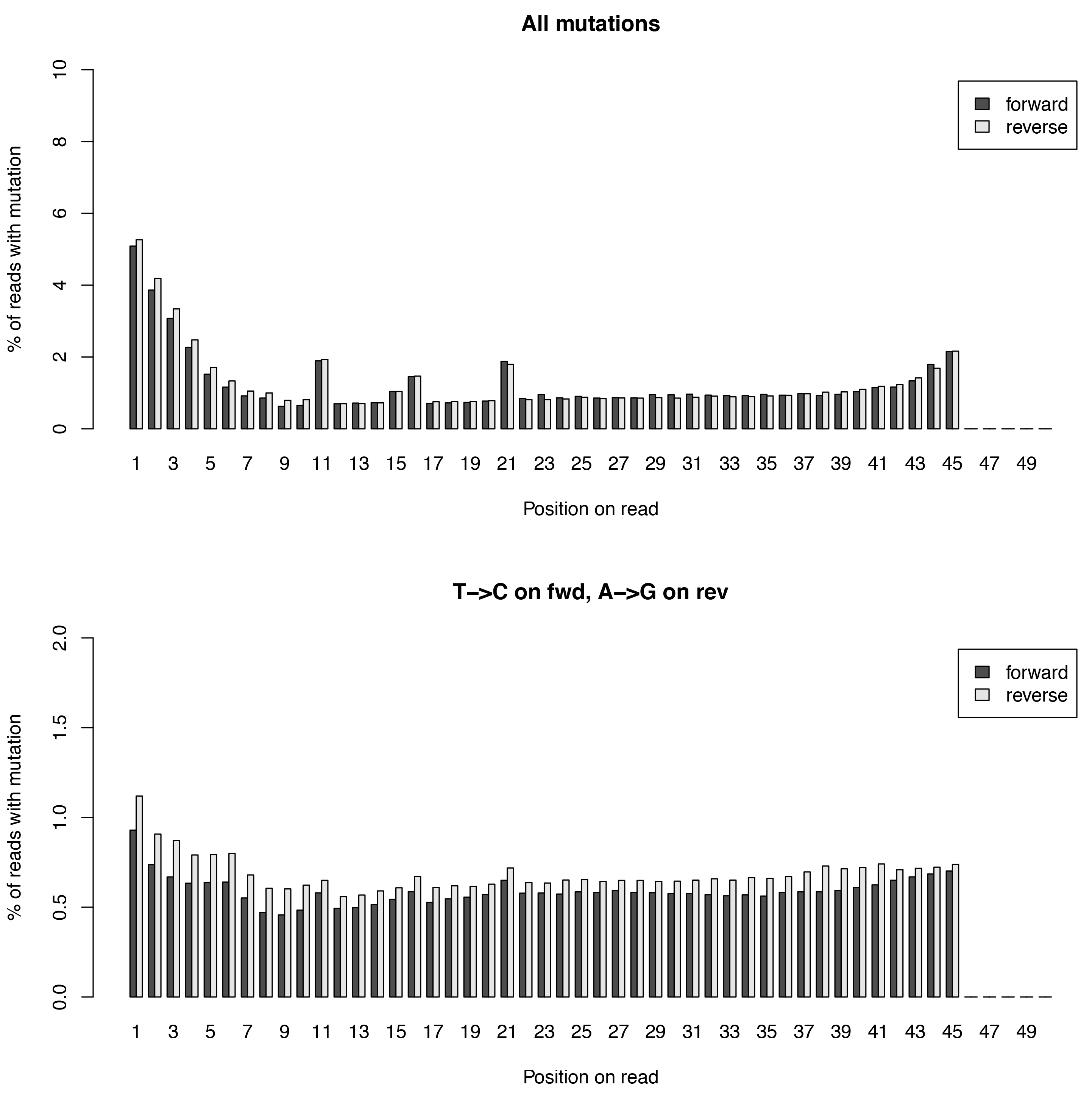
| Parameter | Required | Description |
|---|---|---|
| -h | Prints the help. | |
| -o | x | The output directory where all output files of this tool will be placed. |
| -r | x | The reference fasta file. |
| -mq | Minimum base quality for T->C conversions to be counted. | |
| -t | The number of threads to use. All tools run single-threaded, so it is recommended to use as many threads as available samples. | |
| -s | The called variants from the snp dunk to filter false-positive T->C conversions. | |
| -l | Maximum read length in all samples (will be automatically estimated if not set). | |
| bam | x | BAM file(s) containing the final filtered reads (wildcard * accepted). |
This tool calculates the individual mutation rates per position in an 3’UTR treating T->C mutations separately. This plot can be used to search for biases along UTRs. Only most 3’ 200bp of each UTR will be considered because: * Quantseq fragments are estimated have an average size of ~200bp * This way, any dynamic binning biases can be avoided
alleyoop tcperutrpos [-h] -r <reference fasta> -b <bed file> [-s <SNP directory>]
[-l <maximum read length>] -o <output directory> [-mq <MQ cutoff>]
[-t <threads>] bam [bam ...]
| File | Description |
|---|---|
| Reference fasta | The reference sequence of the genome to map against in fasta format. |
| -s | (optional) The called variants from the snp dunk to filter false-positive T->C conversions. |
| -b | Bed file with coordinates of 3’UTRs. |
| bam | BAM file(s) containing the final filtered reads from slamdunk (wildcard * accepted). |
| File | Description |
|---|---|
| tcperutr.csv | Tab-separated table with mutation rates, one line per UTR position. |
| tcperutr.pdf | Plot of the mutation rates along the UTRs. |
Below is an example plot of mutation rates along all UTRs in a sample. Typically, one will see increasing error rates towards the end of a UTRs. For SLAMseq samples with longer labelling times, the overall T->C conversions in the bottom plot will begin to increase compared to the overall background in the top plot.
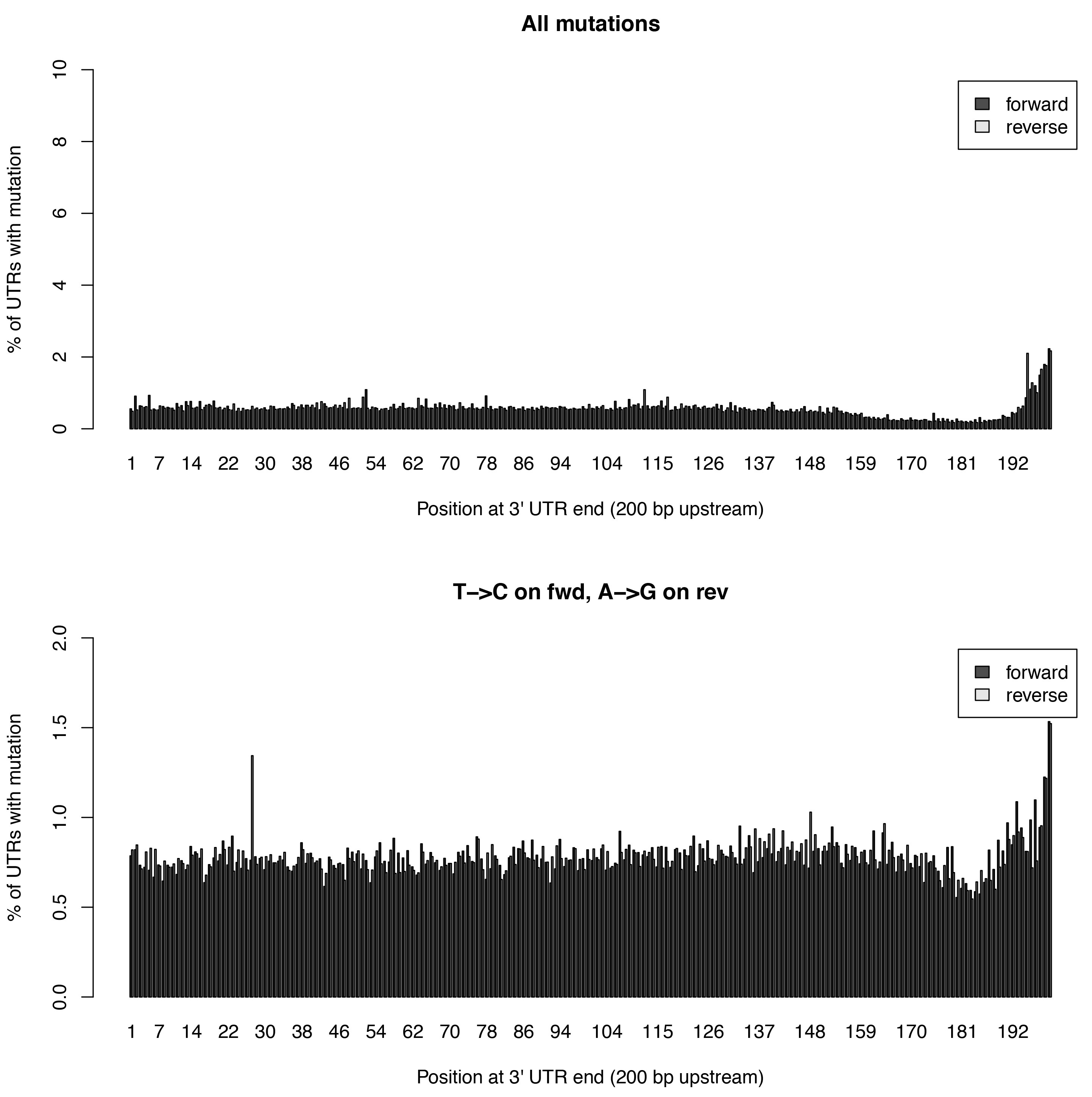
| Parameter | Required | Description |
|---|---|---|
| -h | Prints the help. | |
| -o | x | The output directory where all output files of this tool will be placed. |
| -r | x | The reference fasta file. |
| -b | x | Bed file with coordinates of 3’UTRs. |
| -mq | Minimum base quality for T->C conversions to be counted. | |
| -t | The number of threads to use. All tools run single-threaded, so it is recommended to use as many threads as available samples. | |
| -s | The called variants from the snp dunk to filter false-positive T->C conversions. | |
| -l | Maximum read length in all samples (will be automatically estimated if not set). | |
| bam | x | BAM file(s) containing the final filtered reads (wildcard * accepted). |
This tool outputs all available information calculated by slamdunk for each read in a sample.
alleyoop dump [-h] -r <reference fasta> -s <SNP directory> -o <output directory>
[-mq <MQ cutoff>] [-t <threads>] bam [bam ...]
| File | Description |
|---|---|
| Reference fasta | The reference sequence of the genome to map against in fasta format. |
| -s | The called variants from the snp dunk to filter false-positive T->C conversions. |
| bam | BAM file(s) containing the final filtered reads from slamdunk (wildcard * accepted). |
| File | Description |
|---|---|
| readinfo.sdunk | Tab-separated table with read info, one line per read |
The following columns are contained in the readinfo file:
| Column | Description |
|---|---|
| Name | Name of the read |
| Direction | Read was mapped on forward (1) or reverse (2) strand |
| Sequence | Sequence of the read |
| Mismatches | Number of mismatches in the read |
| tcCount | Number of T->C conversion in the read |
| ConversionRates | List of all possible conversion in the read |
| Parameter | Required | Description |
|---|---|---|
| -h | Prints the help. | |
| -o | x | The output directory where all output files of this tool will be placed. |
| -r | x | The reference fasta file. |
| -mq | Minimum base quality for T->C conversions to be counted. | |
| -t | The number of threads to use. All tools run single-threaded, so it is recommended to use as many threads as available samples. | |
| -s | x | The called variants from the snp dunk to filter false-positive T->C conversions. |
| bam | x | BAM file(s) containing the final filtered reads (wildcard * accepted). |
Docker enables you to run slamdunk out-of-the box wihtout caring about prerequisites and installation procedures. All you need is to install the Docker engine and run the slamdunk Docker image.
You can directly use the slamdunk Docker image from Docker hub .
You can also build them from scratch using any of the two Dockerfiles in slamdunk/docker :
# Build Ubuntu-based image
cd slamdunk/docker/ubuntu
docker build -t <user>/<imagename> .
# View images
docker images
Once the container images is pulled/created, slamdunk can be run out of the box. The only difference is that the data containing directory has to be mounted on the container and all files are relative to that mounting point.
# All data is contained in current working directory and available under /data in the container
docker run -m 8g -v $(pwd):/data tobneu/slamdunk slamdunk map -o /data/map -r /data/GRCm38.fa -5 5 -n 1 /data/testset.fq.gz
Many cloud providers provide Docker support these days. The most prominent cloud provider are the Amazon Web Services (AWS) have a nice integration into Docker machine which easily let’s you install Docker engine on AWS machines. AWS provides free t2.micro instances for free-tier accounts. These only come with 1 GiB of memory which is not enough for mammalian genomes (~ 8 GiB needed) where one would need to go for at least t2.large instances.
# Start a free t2.micro instance (small genomes only e.g. bacteria) on e.g. the us-west-1 region
docker-machine create --driver amazonec2 --amazonec2-region us-west-1 aws-slamdunk
or
# Start a paid t2.large instance (large genomes) on e.g. the us-west-1 region
docker-machine create --driver amazonec2 --amazonec2-region us-west-1 --amazonec2-instance-type t2.large aws-slamdunk
Once the instance has started, one needs to connect to it before running any Docker images.
eval $(docker-machine env aws-slamdunk)
Now everything is set up and one can run slamdunk on an AWS instance.
docker run -m 8g -v $(pwd):/data tobneu/slamdunk slamdunk map -o /data/map -r /data/GRCm38.fa -5 5 -n 1 /data/testset.fq.gz
After the work is done, remember to shut down your AWS instances unless you do not care about paying for an idle instance.
# Shut down
docker-machine stop aws-slamdunk
# Remove
docker-machine rm -y aws-slamdunk
slamdunk is provided under the GNU Affero General Public License v3 .
#!bash
set -x
F=simulation_2/
B=finalAnnotation_test_cut_chrM_correct_100
RL=50
COV=100
SN=pooja_UTR_annotation_examples_sample
rm -rf $F
~/bin/slamdunk/bin/slamsim preparebed -b ${B}.bed -o ${F} -l ${RL}
~/bin/slamdunk/bin/slamsim utrs -b ${F}/${B}_original.bed -o $F -r /project/ngs/philipp/slamseq/ref/GRCm38.fa
~/bin/slamdunk/bin/slamsim reads -b ${F}/${B}_original_utrs.bed -l ${RL} -o $F -cov ${COV} -t 0 --sample-name ${SN}_1_0min
~/bin/slamdunk/bin/slamsim reads -b ${F}/${B}_original_utrs.bed -l ${RL} -o $F -cov ${COV} -t 15 --sample-name ${SN}_2_15min
~/bin/slamdunk/bin/slamsim reads -b ${F}/${B}_original_utrs.bed -l ${RL} -o $F -cov ${COV} -t 30 --sample-name ${SN}_3_30min
~/bin/slamdunk/bin/slamsim reads -b ${F}/${B}_original_utrs.bed -l ${RL} -o $F -cov ${COV} -t 60 --sample-name ${SN}_4_60min
~/bin/slamdunk/bin/slamsim reads -b ${F}/${B}_original_utrs.bed -l ${RL} -o $F -cov ${COV} -t 180 --sample-name ${SN}_5_180min
~/bin/slamdunk/bin/slamsim reads -b ${F}/${B}_original_utrs.bed -l ${RL} -o $F -cov ${COV} -t 360 --sample-name ${SN}_6_360min
~/bin/slamdunk/bin/slamsim reads -b ${F}/${B}_original_utrs.bed -l ${RL} -o $F -cov ${COV} -t 720 --sample-name ${SN}_7_720min
~/bin/slamdunk/bin/slamsim reads -b ${F}/${B}_original_utrs.bed -l ${RL} -o $F -cov ${COV} -t 1440 --sample-name ${SN}_8_1440min
~/bin/slamdunk/bin/slamdunk all -r /project/ngs/philipp/slamseq/ref/GRCm38.fa -b ${F}/${B}_original_utrs.bed -rl 55 -o ${F}/slamdunk ${F}/*.bam
~/bin/slamdunk/bin/slamsim plot.conversions -sim ${F} -slam ${F}/slamdunk/count/ -o ${F}/eval/conversion_rate_eval_plots.pdf
~/bin/slamdunk/bin/slamsim plot.halflifes -sim ${F} -slam ${F}/slamdunk/count/ -t 0,15,30,60,180,360,720,1440 -o ${F}/eval/halflife_per_gene_eval_plots.pdf -b ${F}/${B}_original_utrs.bed
Version 0.3.4
Bugfixes:
Version 0.3.3
Bugfixes:
Version 0.3.2
Changes:
Version 0.3.1
Changes:
Bugfixes:
Version 0.3.0
Changes:
Bugfixes:
Version 0.2.4
Changes:
Bugfixes:
Version 0.2.3
Bugfixes:
Version 0.2.2
Changes:
Bugfixes:
Version 0.2.1
Changes:
Bugfixes:
Version 0.2.0
Due to major changes v0.2.0 is not compatible with v0.1.0. Please rerun alle your samples with slamdunk 0.2.0!
Changes:
Bugfixes :
Version 0.1.0