T>C aware alignment
Using adapted scoring schemes for NextGenMap - a highly sensitive and fast read mapping program - we are able confidently map reads with any reasonable number of T>C conversions to the genome.
SLAMseq is a novel sequencing protocol that directly uncovers 4-thiouridine incorporation events in RNA by high-throughput sequencing. When combined with metabolic labeling protocols, SLAM-seq allows to study the intracellular RNA dynamics, from transcription, RNA processing to RNA stability.
Workflow of SLAMseq
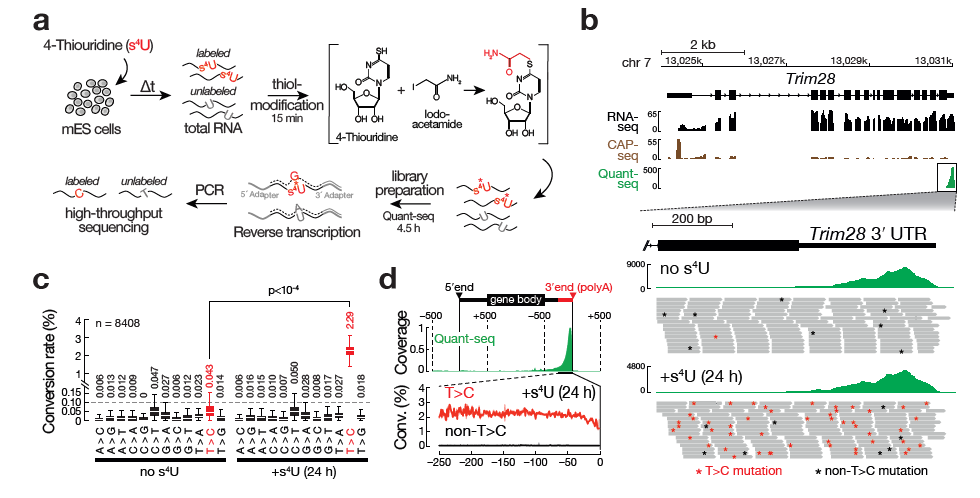
Original publication: Herzog VA et al., Nature Methods, 2017; doi:10.1038/nmeth.4435
pip install slamdunk
conda create --name myenv -c bioconda slamdunk
slamdunk all -r <reference fasta> -b <bed file> -o <output directory> -5 12 -n 100
-t <threads> -m -rl <maximum read length> --skip-sam files [files ...]
This runs slamdunk with its default parameters.
| Parameter | Description |
|---|---|
| -r | The reference fasta file. |
| -b | BED-file containing coordinates for 3’ UTRs. |
| -o | The output directory where all output files of this dunk will be placed. |
| -t | The number of threads to use for this dunk. NextGenMap runs multi-threaded, so it is recommended to use more threads than available samples (default: 1). |
| files | Samplesheet (see Sample file format ) or a list of all sample BAM/FASTA(gz)/FASTQ(gz) files (wildcard * accepted). |
alleyoop utrrates -o <output directory> -r <reference fasta> -t <threads>
-b <bed file> -l <maximum read length> bam [bam ...]
| Parameter | Description |
|---|---|
| -o | The output directory where the plots will be placed. |
| -r | The reference fasta file. |
| -t | The number of threads to use. All tools run single-threaded, so it is recommended to use as many threads as available samples. |
| -b | BED-file containing coordinates for 3’ UTRs. |
| -l | Maximum read length in all samples. |
| bam | BAM file(s) containing the final filtered reads from the filter folder (wildcard * accepted). |
SlamDunk consists of 4 core modules to process SLAMeq data:
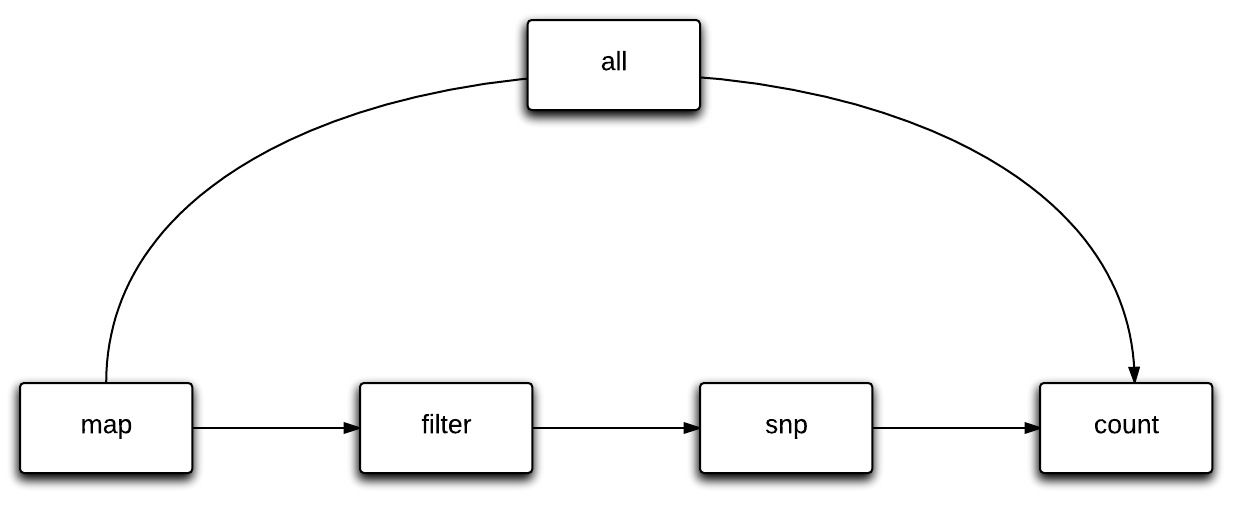
Using adapted scoring schemes for NextGenMap - a highly sensitive and fast read mapping program - we are able confidently map reads with any reasonable number of T>C conversions to the genome.
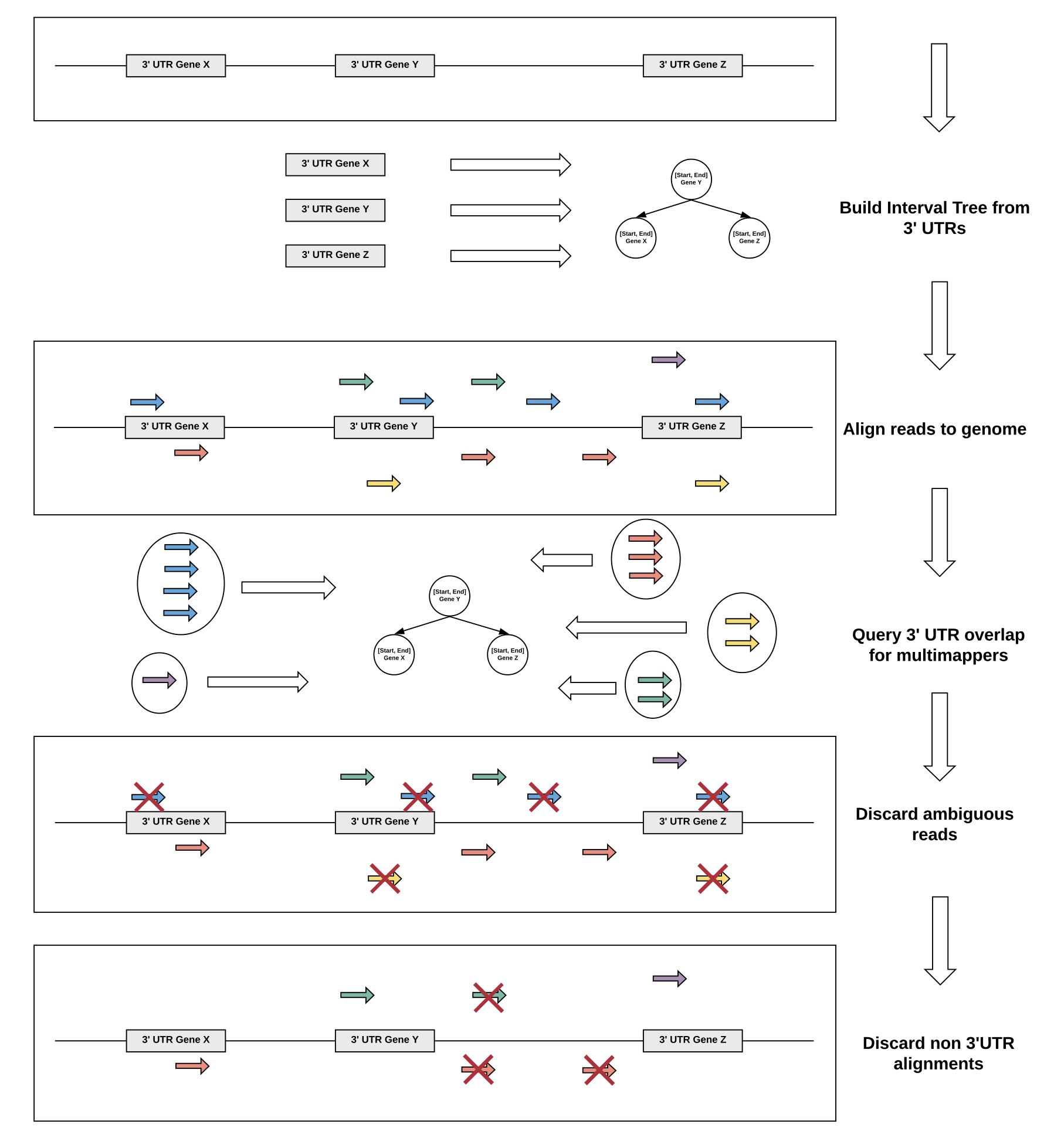
We devised a reference guided approach to utilize multimapper information in 3'UTR regions of low complexity using efficient data structures.
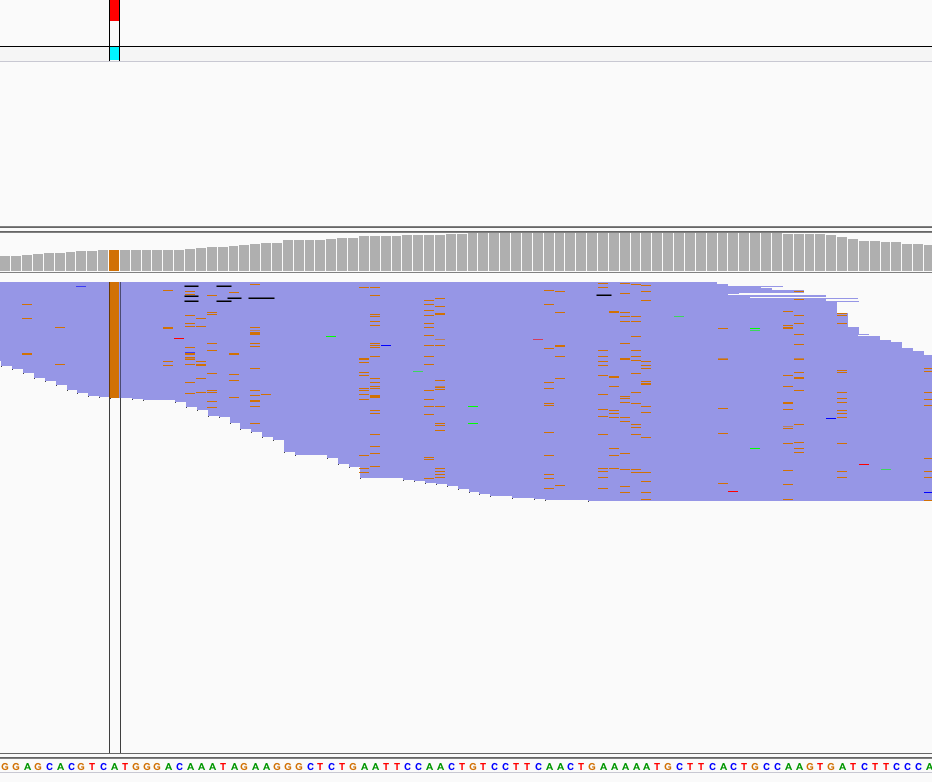
Using Varscan2 we can separate true T>C conversions from false-positives caused by variants.
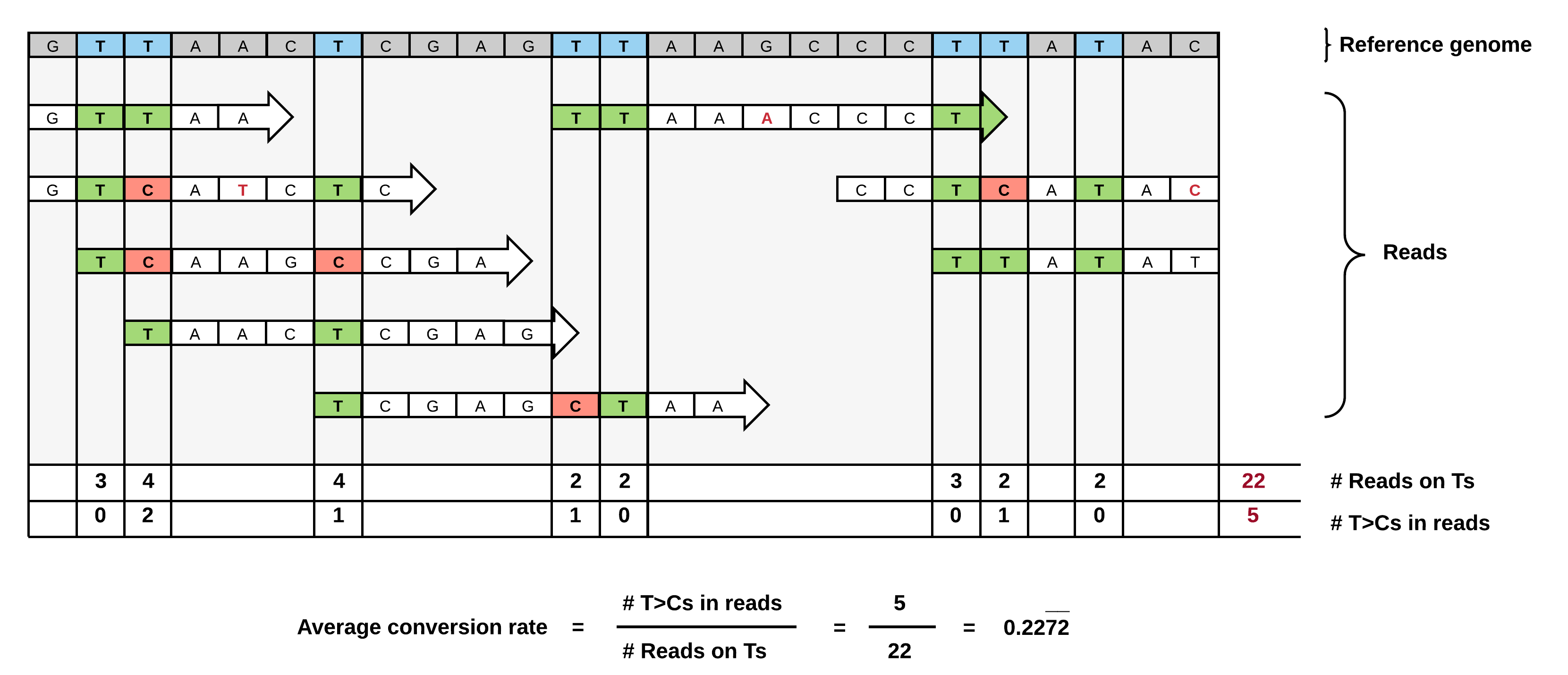
We devised a conversion counting method normalizing for T-content and coverage in individual 3'UTRs for unbiased comparison of transcripts.
slamdunk depends on several python packages listed in the requirements.txt file.
In addition, slamdunk uses the following external software for its analysis:
Both alleyoop and splash require R (v3.2.2). All R package dependencies are resolved automatically from CRAN during installation.
SlamDunk is hosted as Python package on the Python Package Index. You can install it with a single command using pip.
# Root
pip install slamdunk
# Local user
pip install --user slamdunkFor maximum convenience, you might also consider running slamdunk using our Docker image.
MultiQC is a popular tool to aggregate results from bioinformatics analyses across many samples into a single report.
We implemented a MultiQC module which was released with MultiQC v0.9 to support integration of SlamDunk QC plots and statistics into your MultiQC reports.
Currently we support integration of the summary, rates, utrrates, tcperreadpos and tcperutrpos modules.
You can download this report and / or the logs used to generate it, to try running MultiQC yourself.
Many thanks to Phil Ewels for his support!
Veronika A. Herzog, Brian Reichholf, Tobias Neumann, Philipp Rescheneder, Pooja Bhat, Thomas R. Burkard, Wiebke Wlotzka, Arndt von Haeseler, Johannes Zuber & Stefan L. Ameres.
Nature Methods, 2017, http://doi.org/10.1038/nmeth.4435
Matthias Muhar, Anja Ebert, Tobias Neumann, Christian Umkehrer, Julian Jude, Corinna Wieshofer, Philipp Rescheneder, Jesse J. Lipp, Veronika A. Herzog, Brian Reichholf, David A. Cisneros, Thomas Hoffmann, Moritz F. Schlapansky, Pooja Bhat, Arndt von Haeseler, Thomas Köcher, Anna C. Obenauf, Johannes Popow, Stefan L. Ameres & Johannes Zuber.
Science, 2018, http://doi.org/10.1126/science.aao2793
Wayo Matsushima, Veronika A. Herzog, Tobias Neumann, Katharina Gapp, Johannes Zuber, Stefan L. Ameres, Eric A. Miska.
Development, 2018, http://doi.org/10.1242/dev.164640
Neumann, T., Herzog, V. A., Muhar, M., Haeseler, von, A., Zuber, J., Ameres, S. L., & Rescheneder, P. (2019). Quantification of experimentally induced nucleotide conversions in high-throughput sequencing datasets. BMC Bioinformatics, 20(1), 258. http://doi.org/10.1186/s12859-019-2849-7
SlamDunk is developed by Tobias Neumann and Philipp Rescheneder.