ICLR 2025 digest
This April, I had the opportunity to attend the International Conference on Learning Representations (ICLR) 2025 in Singapore (after missing the 2024 edition in Vienna, shame on me). ICLR has established itself as the premier gathering for professionals dedicated to representation learning and deep learning, and I needed to know what the fuzz was about.
The conference brought together an impressive mix of academic researchers, industry practitioners from companies like Google DeepMind, Meta, and Isomorphic Labs, as well as entrepreneurs and graduate students - all converging on Singapore to discuss cutting-edge research spanning machine vision, computational biology, speech recognition, and robotics.
Beyond the technical content, Singapore itself provided a stunning backdrop. The tropical climate, lush greenery, and modern architecture - especially the iconic Marina Bay Sands - created an very futuristic atmosphere. The city food scene and details like subway pop choreography performances added to the experience. But let me dive into what mattered most to me: the technical developments that are shaping the future of AI.

The Rise of Agentic AI
If there was one overarching theme at ICLR 2025, it was the evolution of AI systems from passive responders to active agents. The shift from traditional deep learning models to agentic AI represents a fundamental change in the field of artificial intelligence.
From Models to Agents
Traditional AI systems, even sophisticated ones, essentially function as input-output machines. You provide data, they provide predictions. Agentic AI systems, by contrast, exhibit several key characteristics that make them fundamentally different:
Goal-oriented behavior: Rather than simply responding to prompts, agentic systems can pursue complex, multi-step objectives autonomously. They don’t just answer “what should I do next?” - they actually do it.
Reflection and adaptation: Perhaps most intriguingly, these systems can reflect upon their own work and iteratively improve their approach. This meta-cognitive capability allows them to identify failures, adjust strategies, and come up with follow-up steps without human intervention.
Environment interaction: Agentic AI systems actively interact with their environment - whether that’s a database, a laboratory instrument, or a computational workflow. They can query information, execute commands, and observe the results of their actions.
Tool integration: Modern agentic systems seamlessly integrate with external tools and APIs, allowing them to leverage specialized capabilities beyond their core model. For instance, TxAgent - a therapeutic reasoning agent presented at the conference - orchestrates 211 specialized tools spanning FDA drug databases, Open Targets, and the Human Phenotype Ontology. This dynamic tool selection allows agents to access verified, continually updated knowledge rather than relying solely on their training data.
Multi-agent collaboration: Finally and most futuristic, these systems can coordinate with other AI agents, dividing tasks and sharing information to solve problems that would be intractable for a single agent.
Several talks at ICLR covered agentic systems targetting complex scientific workflows, from literature review and hypothesis generation to experimental design and data analysis. My favourite showcase was Agentic-Tx, a therapeutics-focused system powered by Gemini 2.5, which achieved a 52.3% relative improvement over o3-mini on Humanity’s Last Exam (Chemistry & Biology) and demonstrated significant gains on ChemBench and GPQA benchmarks. The key insight is that these systems aren’t just faster versions of traditional AI - they represent a qualitatively different approach to automation.
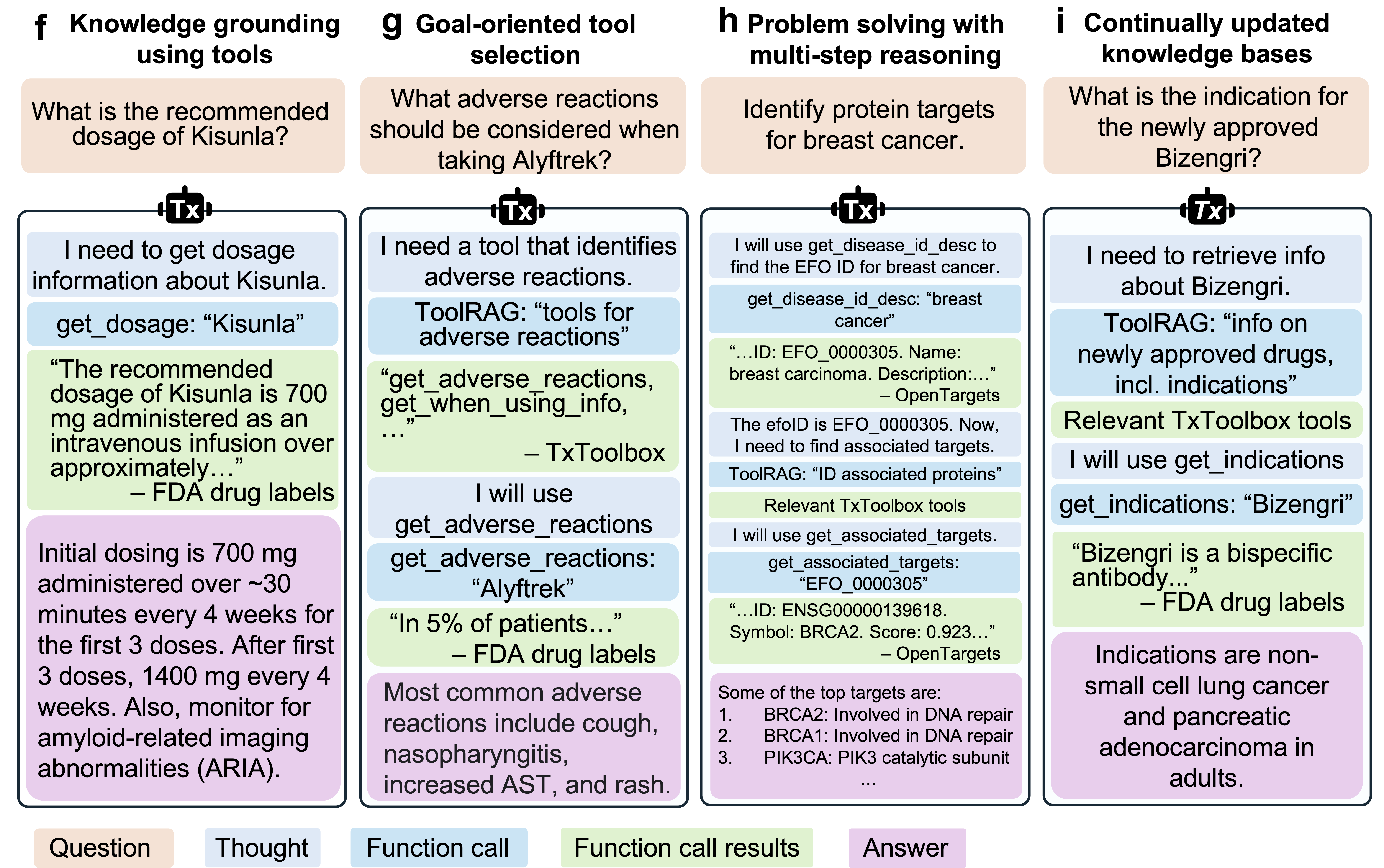
TxAgent workflow demonstrating agentic AI capabilities: knowledge grounding through tool calls, goal-oriented tool selection, multi-step reasoning, and access to continuously updated knowledge bases. The system generates transparent reasoning traces that show each decision step. Source: Gao et al., 2025
TxGemma: A Case Study in Domain-Specific Agents
The most interesting development coming from a drug discovery company was TxGemma, a suite of efficient, domain-specific large language models for therapeutic applications. What makes TxGemma noteworthy isn’t its performance, but its practical accessibility and adaptability for drug discovery.
Built on the Gemma-2 architecture, TxGemma comes in three sizes - 2B, 9B, and 27B parameters - making it dramatically more efficient than typical foundation models. The suite was fine-tuned on a comprehensive dataset of 7.08 million training samples from the Therapeutics Data Commons (TDC), covering 66 different therapeutic development tasks spanning small molecules, proteins, nucleic acids, diseases, and cell lines. This broad training enables TxGemma to handle diverse aspects of drug discovery, from early-stage target identification to late-stage clinical trial predictions.
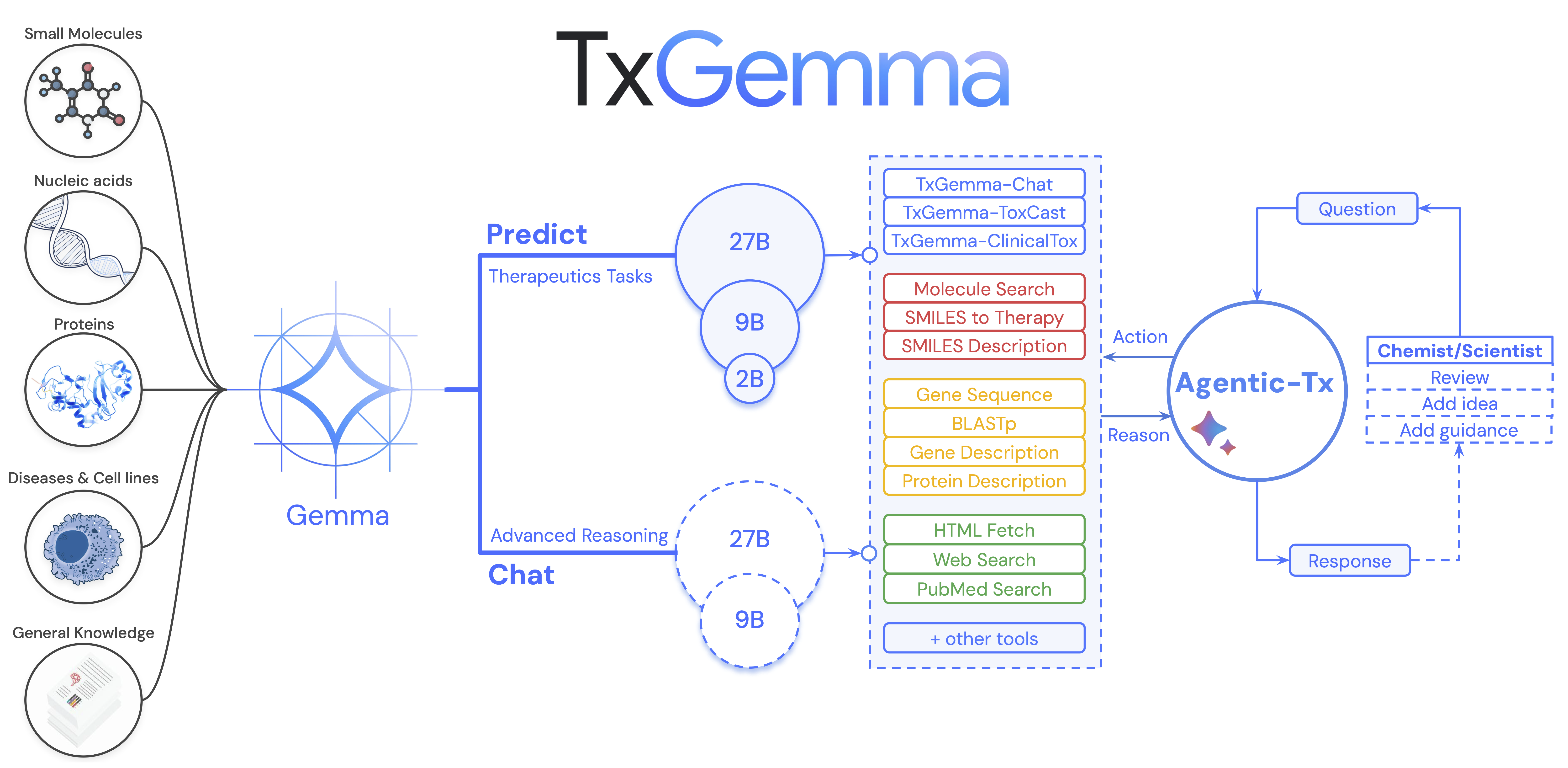
TxGemma model family: Three size variants (2B, 9B, 27B) trained on diverse therapeutic data from TDC, with specialized versions for prediction (TxGemma-Predict) and conversation (TxGemma-Chat). The models can be integrated as tools in agentic systems like Agentic-Tx. Source: Wang et al., 2025
The performance results very definitely useful: Across the 66 TDC tasks, TxGemma achieved superior or comparable performance to state-of-the-art models on 64 tasks (outperforming on 45), despite being orders of magnitude smaller than many competing models. On tasks involving drug-target interactions, pharmacokinetics, and toxicity prediction, TxGemma consistently matched or exceeded specialist models that were designed specifically for those narrow applications.
What’s particularly intriguing is TxGemma’s data efficiency. When fine-tuning for clinical trial adverse event prediction, TxGemma matched the performance of base Gemma-2 models using less than 10% of the training data. In data-scarce domains like drug discovery - where proprietary datasets are common and expensive to generate - this efficiency advantage is definitely a major plus if you want to put this in production.
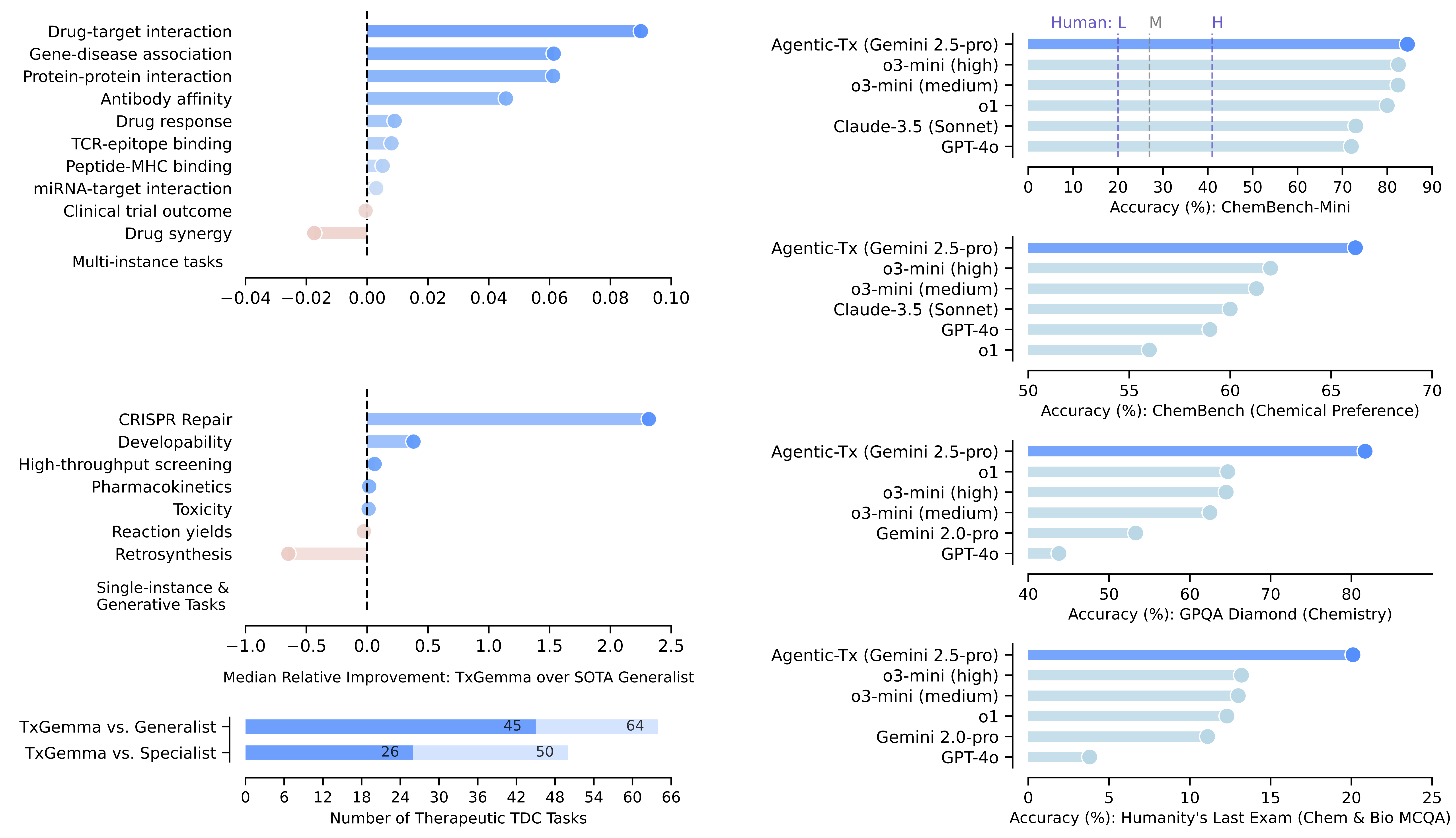
TxGemma-Predict demonstrates superior performance across diverse therapeutic task types, with particularly strong results on multi-instance tasks involving multiple data modalities. Median relative improvements show consistent gains over both generalist and specialist state-of-the-art models. Source: Wang et al., 2025
The real power of domain-specific foundational models lies in their ability to serve as starting points. Rather than training from scratch, researchers can fine-tune TxGemma on their specific tasks, dramatically reducing the computational resources and data required to achieve good performance. The models can even run on a single Nvidia H100 GPU, making them accessible to smaller research groups and enabling local deployment for sensitive applications.
TxGemma had also a quite nice illustrative example how it can be used in Agentic Systems - theirs being Agentic-Tx, a therapeutics-focused agentic system powered by Gemini 2.5 that extends TxGemma’s capabilities by orchestrating complex workflows. Agentic-Tx employs a modular, tool-usage paradigm, in contrast to TxGemma’s direct generation of solutions. Agentic-Tx utilizes the ReAct framework, allowing it to interleave reasoning steps (“thoughts”) with actions (tool use). The agentic system receives a task or question and iteratively takes actions based on its current context and therefore answer questions that involve multiple reasoning steps to solve, for example “What structural modifications could improve the potency of the given drug?” requires iteratively searching the drug’s structural space and then prompting TxGemma to predict potency.
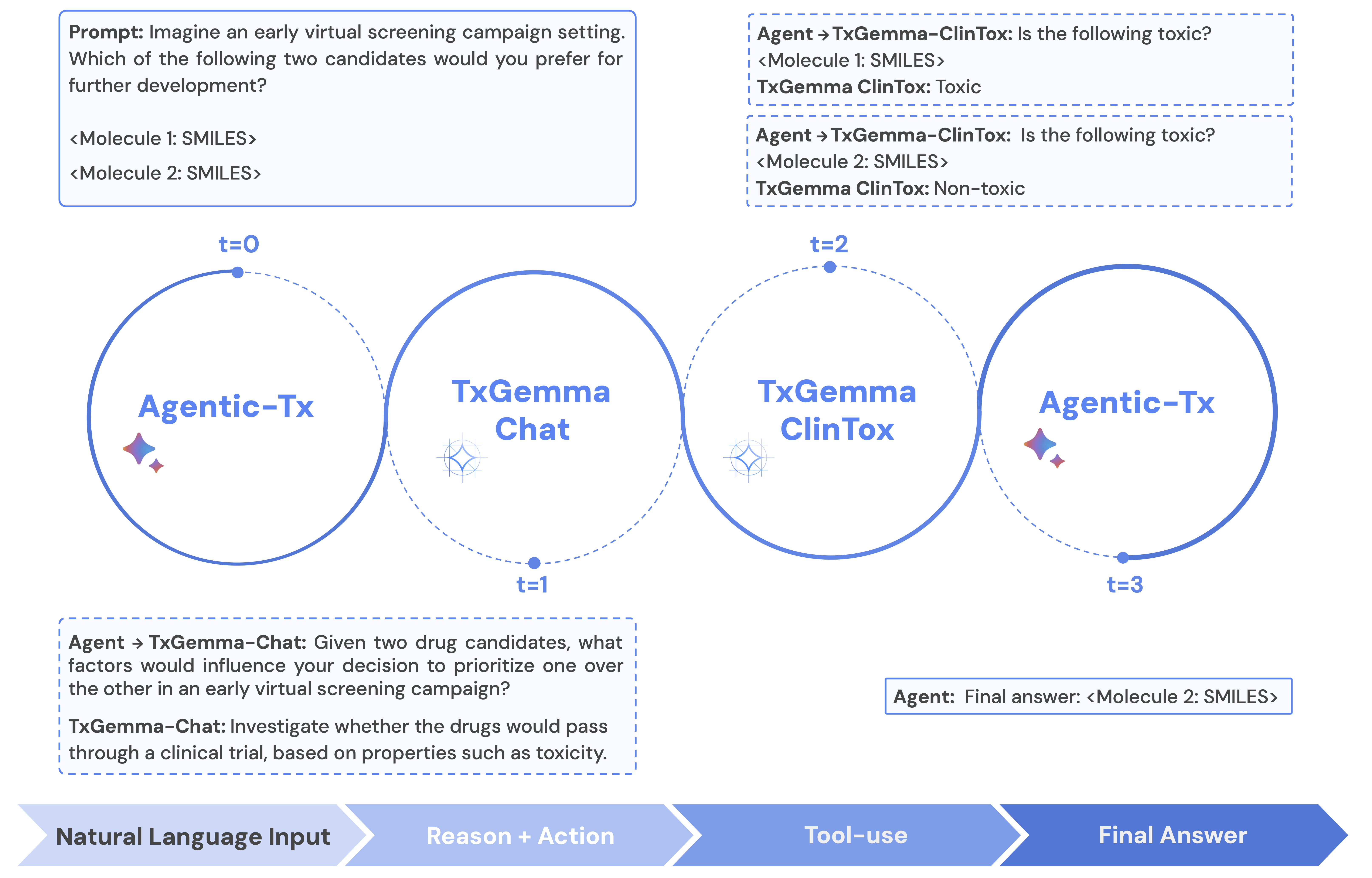
Agentic-Tx in combination with the ReAct framework to interleave thought with tool-usage. In this example, Agentic-Tx uses two tools to prioritize which hit from a screening campaign should be prioritized: TxGemma-Chat and the clinical toxicity prediction tool based on TxGemma-Predict.
Lastly, TxGemma goes beyond prediction. Unlike traditional models that output only answers, TxGemma-Chat - the conversational variant - can explain its reasoning. When asked why a molecule crosses the blood-brain barrier, it can discuss lipophilicity, molecular weight, and hydrogen bonding based directly on the molecular structure. This explainability is a first in therapeutic AI and addresses one of the field’s most significant limitations: the “black box” problem.
TxGemma-Chat maintains this conversational ability while accepting only about a 10% performance reduction on predictive tasks compared to TxGemma-Predict. This trade-off - slightly lower raw accuracy for vastly improved interpretability and user interaction - represents an important design decision in therapeutic AI. For research applications where understanding model reasoning is crucial to get more insights into relevant decicsive parameters, this tradeoff is definitely negligible.
Moreover, TxGemma has been released as an open model specifically trained only on commercially licensed datasets. This decision recognizes the prevalence of proprietary data in pharmaceutical research and allows smaller biotech pharmaceutical startups to adapt and validate the models on their own datasets, potentially tailoring performance to their specific research needs and real-world applications.
Multimodal Learning: Connecting Different Data Types
Another major theme at ICLR was the challenge of integrating different types of biological and chemical data. In drug discovery and computational biology, we often have rich datasets in different modalities - transcriptomics, proteomics, chemical structures, microscopy images - but connecting these disparate data types is challenging.
Multimodal Adapters: Efficient Cross-Modal Learning
One elegant solution presented at the conference was the concept of multimodal adapters - specifically, the single-cell Drug-Conditional Adapter (scDCA). The core idea is pretty simple: rather than training massive end-to-end models that try to handle all modalities simultaneously, we can train small “adapter” layers that bridge between pre-trained foundational models for each modality.
Here’s how it works. You might have a powerful foundational model for single-cell transcriptomics (like scGPT, trained on 33 million cells) and another for molecular structures (like ChemBERTa, trained on 77 million compounds). Rather than starting from scratch to predict how drugs affect cells, scDCA introduces lightweight adapter layers that learn to translate molecular information into adjustments of the cellular model’s internal representations.
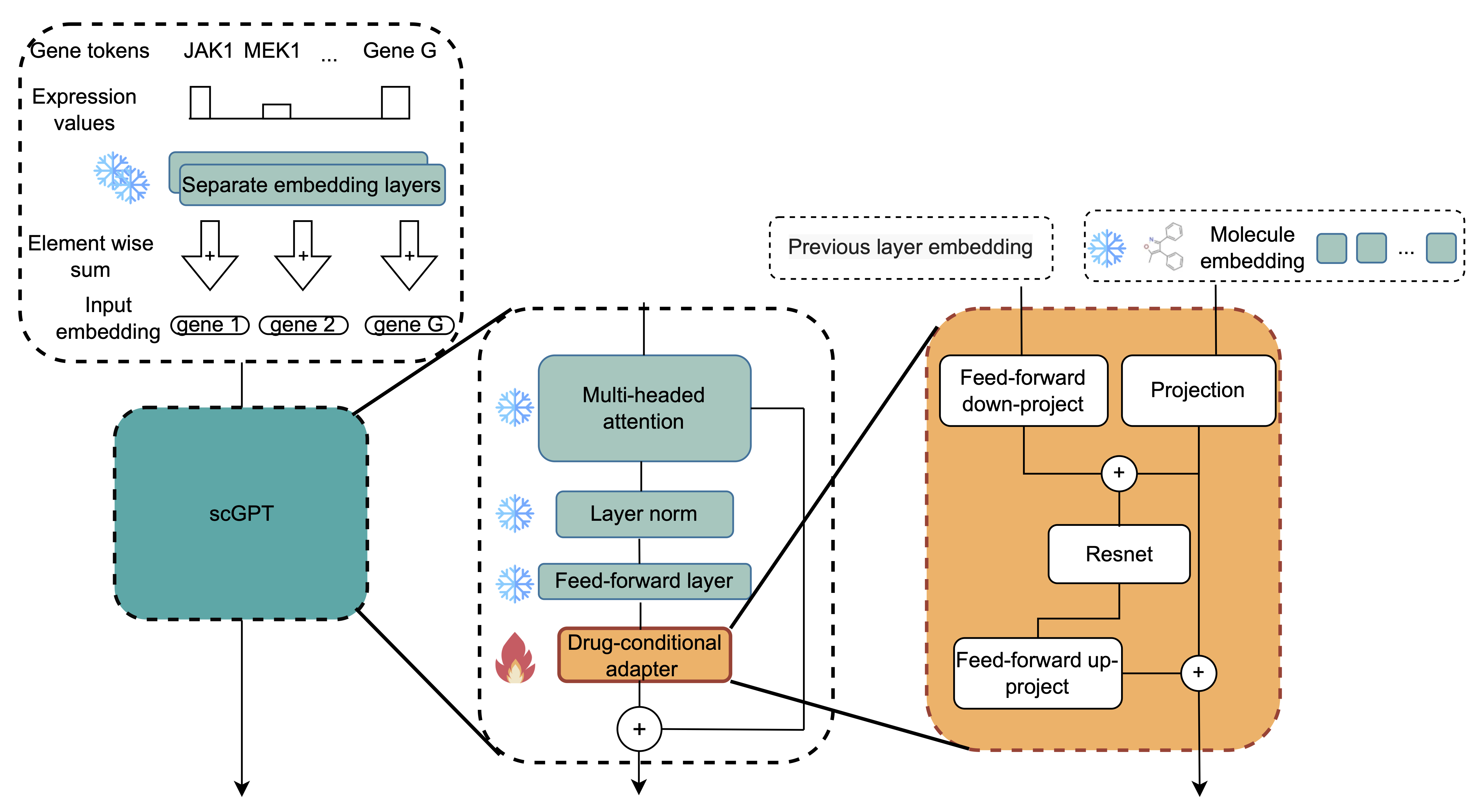
Architecture of scDCA showing drug-conditional adapters that efficiently fine-tune single-cell foundation models. The adapter introduces molecular conditioning through dynamic bias adjustments while keeping the original transformer weights frozen, enabling training with less than 1% of the original model’s parameters. Source: Maleki et al., 2025
The advantages are the pretty striking:
Efficiency: Adapters typically involve training only 1% of the parameters compared to the original foundational models. For scDCA specifically, while the base scGPT model has millions of parameters, the adapters add only a tiny fraction. This makes them dramatically faster and cheaper to train - critical when working with limited datasets.
Avoiding overfitting: By keeping the foundational models frozen and only training the adapter, you preserve the learned knowledge in the original models. This is particularly valuable when working with limited paired training data - a common challenge when you have only 188 compounds with cellular response data (as in the Sciplex3 dataset used for validation).
Flexibility: You can mix and match different foundational models by simply training new adapters, without needing to retrain entire systems. Need to connect a different molecular encoder? Just train a new adapter layer.
Performance was pretty neat: scDCA successfully predicted cellular responses to novel drugs and - even more impressively - generalized to completely unseen cell lines in a zero-shot setting with 82% accuracy. This generalization happens because the frozen single-cell foundation model supposedly retains its understanding of gene-gene interactions and cellular states, while the adapter learns to modulate these representations based on molecular structure.
Multimodal Lego: Assembling Models Like Building Blocks
Taking the adapter concept even further, MM-Lego (Multimodal Lego) introduced a framework that makes any set of encoders compatible for model merging and fusion - without requiring paired training data at all.
The key innovation is the “LegoBlock” - a wrapper that enforces two critical properties. First, it ensures all modalities produce latent representations with the same dimensions (making them stackable, like Lego pieces). Second, and more cleverly, it learns representations in the frequency domain using Fourier transforms. Why does this matter? Frequency-domain representations apparently are less prone to signal interference when combined, making them ideal for model merging (I have to trust them on this ^^).
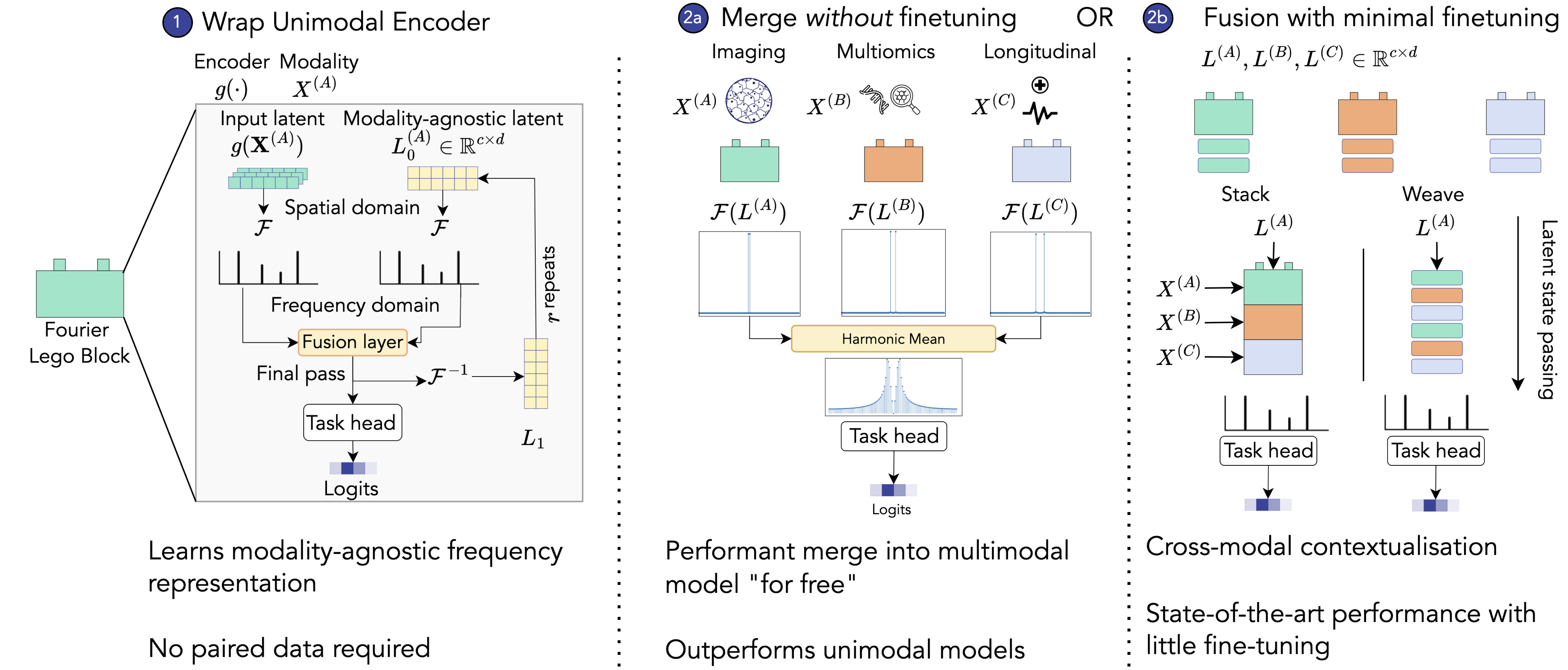
The MM-Lego workflow showing how LegoBlocks enforce structural compatibility and learn frequency-domain representations that enable merging without signal interference. Models can be merged without any fine-tuning (LegoMerge) or with minimal fine-tuning for state-of-the-art performance (LegoFuse). Source: Hemker et al., 2024
MM-Lego introduced two approaches:
LegoMerge: Combines models trained entirely separately - without any paired data or fine-tuning. The merged representation uses a harmonic mean of magnitudes and arithmetic mean of phases in the frequency domain, carefully designed to avoid one modality dominating the signal. Interestingly and counter-intuitively, this achieved competitive performance with end-to-end trained models across seven medical datasets, despite never seeing a single multimodal training sample.
LegoFuse: Takes the merged components and fine-tunes them for just a few epochs (as little as 2) with paired data. This allows modalities to mutually contextualize each other while avoiding the computational overhead of full end-to-end training. LegoFuse achieved state-of-the-art results on 5 of 7 benchmarked tasks.
The practical advantages are substantial: MM-Lego scales linearly with the number of modalities (not quadratically like many attention-based methods), handles missing modalities gracefully, works with non-overlapping training sets, and - critically - doesn’t require architecturally identical models. You can combine a CNN for images with a transformer for sequences simply by wrapping each in a LegoBlock.
One presentation demonstrated training on completely non-overlapping datasets - one set of patients with histopathology slides, a different set with genomic data, both with the same clinical outcomes. Traditional end-to-end models can’t handle this scenario at all, but MM-Lego achieved strong performance by training each modality independently and merging the results.
Why This Matters
These approaches address fundamental challenges we face everyday in computational biology and drug discovery. Paired multi-modal measurements are expensive and often impossible - you might have single-cell data for some conditions and bulk sequencing for others, microscopy for some samples and proteomics for different samples. Traditional methods force you to either throw away data (using only the intersection) or impute missing values (introducing noise).
Adapter-based and frequency-domain approaches like scDCA and MM-Lego let you leverage all available data by training on unpaired samples and combining models afterward. As Michael Bronstein memorably put it in his panel discussion: “Everybody wants to develop the next AlphaFold, nobody the next PDB.” The bottleneck isn’t model architecture - it’s generating high-quality data at scale. Methods that work with incomplete, unpaired, and heterogeneous data are essential for making progress.
Contrastive Learning: Learning from Similarity
Contrastive learning was also another recurring topic at ICLR, particularly for biological and chemical applications where labeled data can be scarce but unlabeled structure is abundant.
The Core Principle
The fundamental idea behind contrastive learning is elegantly simple: teach a model to recognize what’s similar and what’s different. Rather than requiring explicit labels for every example, you create pairs of data points and ask the model to learn that similar pairs should have similar representations, while dissimilar pairs should be far apart in representation space.
In the context of biological perturbations, this might mean:
- Similar pairs: Unperturbed samples from the same cell line, or cells treated with the same compound
- Dissimilar pairs: Perturbed vs. unperturbed samples, or cells treated with different compounds
By training models to maximize agreement within similar pairs and maximize disagreement between dissimilar pairs, you can learn rich representations that capture meaningful biological variation.
Better Separation, Better Biology
Several presentations demonstrated how contrastive learning leads to better separation of perturbations in embedding spaces. This has practical implications for downstream analyses - better UMAPs, clearer clustering, and more interpretable representations of complex biological states.
One particularly clever application involved using different molecular representations (SMILES strings, graphs, 3D conformations) and creating contrastive pairs based on their chemical similarity. Despite working with relatively small domain-specific datasets (1.5 million compounds), this approach produced models with performance comparable to those trained on billions of general chemical structures.
The key insight is that contrastive learning allows you to leverage the structure inherent in your data - the relationships between samples - rather than requiring expensive manual annotations for every data point.
Beyond One-Dimensional Learning: LangPert and Hybrid LLM Approaches
Several talks pushed contrastive learning into more sophisticated territory, combining it with Large Language Models to predict unseen perturbations. One particularly innovative approach was LangPert, presented by researchers from Novo Nordisk, which demonstrates a clever way to leverage LLMs’ biological knowledge without falling victim to their numerical limitations.
The core challenge is predicting cellular responses to genetic perturbations you’ve never experimentally tested. Traditional foundation models like scGPT and graph neural networks like GEARS have tackled this, but as we have seen repeatedly, even sophisticated deep learning methods often struggle to beat simple baselines like predicting mean expression.
LangPert’s insight is elegantly simple: let the LLM do biological reasoning, and let traditional methods handle the numbers. LLMs have absorbed vast scientific literature and “know” about gene functions, pathways, and interactions. But they’re terrible at handling high-dimensional gene expression data - thousands of numerical values that would choke on tokenization alone.
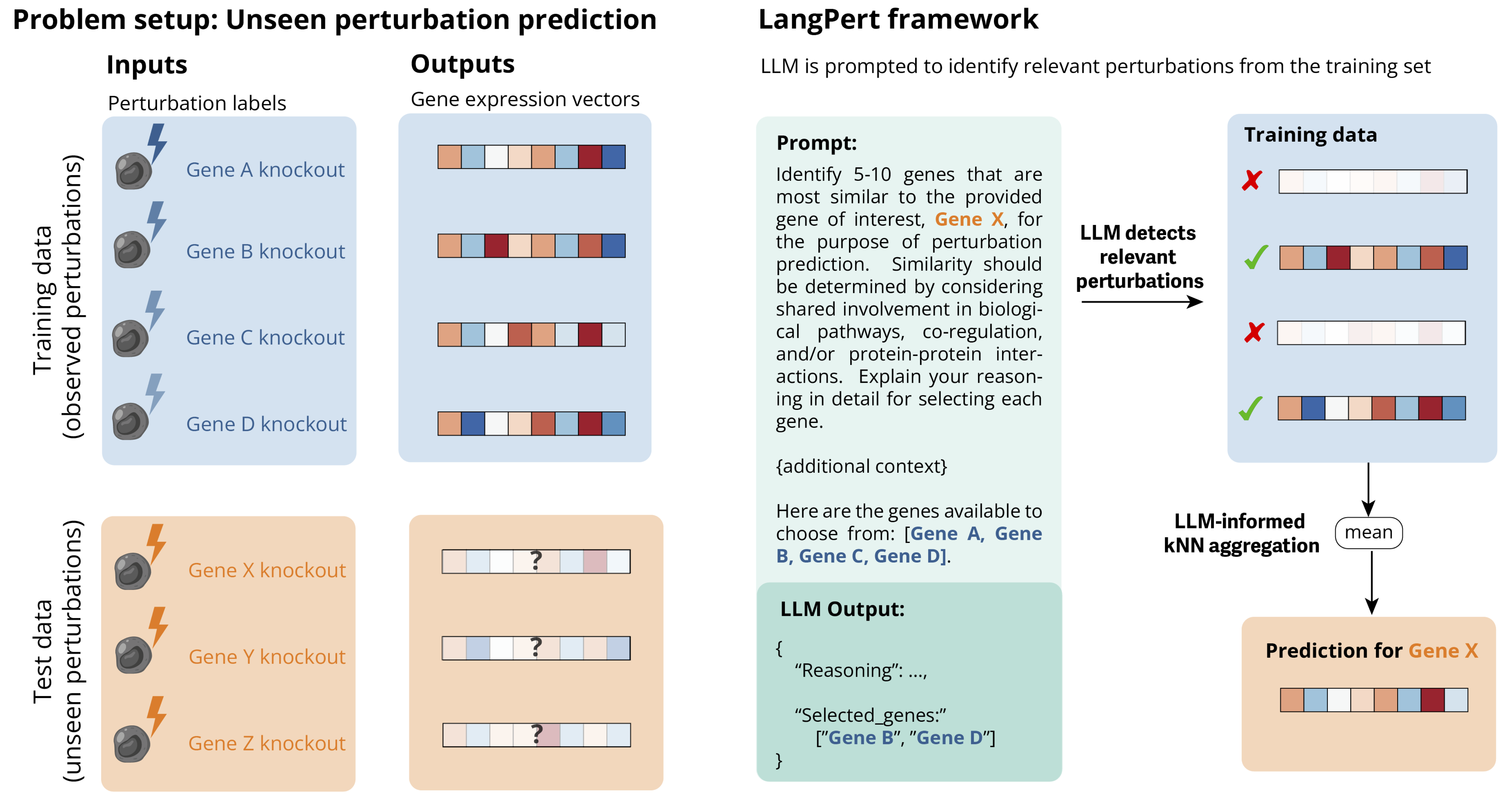
The LangPert framework architecture: Instead of asking LLMs to directly predict high-dimensional gene expression vectors, the system leverages LLMs to identify biologically relevant training examples. For an unseen perturbation (x), the LLM examines all available training perturbations and selects a small subset of functionally related genes. These LLM-selected examples then guide a k-nearest neighbors aggregator that performs the actual numerical prediction in the high-dimensional expression space. This hybrid approach combines the biological reasoning capabilities of LLMs with efficient numerical computation. Source: Märtens et al., 2025*
The framework works in two steps. First, when predicting the effects of an unseen gene knockout, LangPert asks the LLM: “Which genes from my training set are most functionally similar to this target gene?” For instance, if predicting SMG5 (involved in mRNA decay), the LLM might select UPF1, UPF2, and RBM8A - all core components of the same pathway. The LLM provides biological reasoning: these genes participate in similar cellular processes and likely produce similar knockout effects.
Second, the system simply averages the actual experimental expression profiles of these LLM-selected genes. This is k-nearest neighbors with a twist - the “neighbors” are chosen by biological reasoning rather than numerical distance in expression space.
On the K562 benchmark, LangPert achieved substantially better performance than previous methods, and this advantage held across different data regimes. What makes it work? Unlike static embeddings, LangPert dynamically reasons about relevance for each prediction. Different frontier LLMs (Claude, OpenAI o1, o3-mini) select somewhat different gene sets yet achieve similar performance, suggesting multiple valid biological paths to good predictions. The system can even incorporate self-critique, asking the LLM to refine its initial selections.
This “LLM-informed contextual synthesis” represents a template for integrating LLMs into scientific workflows more broadly. Rather than forcing LLMs to handle everything, we architect systems where LLMs do conceptual reasoning while traditional methods handle precise numerical operations. The approach also maintains interpretability - you can examine which genes were selected and read the LLM’s biological rationale, crucial for scientific applications where understanding why matters as much as the predictions themselves.
Training Strategies and Data Quality
Amidst all the excitement about new architectures and approaches, several sobering talks reminded attendees about fundamental challenges in model training and evaluation.
The Train-Test Split Problem
One eye-opening presentation highlighted a subtle but critical issue in molecular machine learning: how we split our data for training and testing. The standard approach of random splitting can lead to severely imbalanced distributions of molecular similarity between training and test sets.
Here’s why this matters: if your test set happens to contain many molecules very similar to your training set, your model will appear to perform much better than it actually does. The good performance on easy (similar) test examples masks poor performance on truly novel molecules.
The proposed solution - Similarity-Aware Evaluation (SAE) - explicitly controls the distribution of similarities in test sets to ensure balanced evaluation. This gives a more honest assessment of how well models generalize to genuinely new chemical space.
This serves as a reminder that methodological rigor in evaluation is just as important as sophisticated model architectures. Without proper evaluation strategies, we risk fooling ourselves about our models’ capabilities.
The Data Generation Bottleneck
Perhaps my most memorable quote from the conference came from Michael Bronstein during a panel discussion: “Everybody wants to develop the next AlphaFold, nobody wants to develop the next Protein Data Bank.”
This pithy observation captures a fundamental tension in computational biology and drug discovery. We’re incredibly good at building sophisticated AI models, but generating the high-quality, large-scale datasets these models need remains a major bottleneck.
Several speakers from both academia and industry emphasized this point. When I chatted with Google DeepMind , they also mentioned struggling with throughput in their lab automation efforts - testing only 20-30 proteins every two weeks. Meanwhile, unlearning and safety talks highlighted that even with massive datasets, ensuring data quality and removing harmful content remains challenging.
The message to me was clear and also somewhat encouraging when facing behemoths like OpenAI: the next major advances in AI for science won’t come from slightly better architectures, but from systematic approaches to generating better data at scale.
Industry Perspectives
One of the highlights of attending ICLR was the opportunity to mingle with researchers from major industry players. Networking events at Marina Bay Sands brought together people from Google DeepMind, Meta, Isomorphic Labs, and various biotech companies.
Isomorphic Labs
Isomorphic Labs, built around the core IP of AlphaFold3, has made impressive progress in translating academic breakthroughs into practical drug discovery. Their partnerships with Novartis and Eli Lilly (totaling over $90M in upfront payments, with a $700M investment round) signal serious industry confidence.
What’s particularly interesting is their ambition to apply AI across the entire drug development process, not just structure prediction. This includes ADME (absorption, distribution, metabolism, and excretion) prediction - traditionally challenging areas that have resisted computational approaches and I have huge reservations that they can win on this turf, as big pharma players already have massive proprietary datasets in their hands.
According to conversations at the conference, access to internal pharmaceutical ADME datasets from their partners could be a game-changer, potentially allowing them to train models on data that has never been publicly available.
Google DeepMind: Building the Lab-in-the-Loop
DeepMind’s research arm is taking a complementary approach, focusing on protein design and active learning with automated laboratories. Their vision of a fully automated “lab-in-the-loop” system - where AI designs experiments, robots execute them, and the results feed back to improve the AI - remains aspirational but compelling.
Interestingly, they maintain relatively small labs with simple readouts (protein binding, basic toxicity assays), suggesting that even with Google’s resources, scaling experimental throughput remains challenging. This again reinforces the data generation bottleneck theme.
Relevant Data Resources
Throughout the conference, several valuable data resources were repeatedly mentioned. These public datasets are enabling the current wave of AI applications in biology and chemistry:
Therapeutics Data Commons (TDC): A comprehensive collection of datasets for therapeutic applications, spanning from molecular properties to clinical outcomes.
PrimeKG: A holistic knowledge graph integrating 20 high-quality biomedical resources, describing over 17,000 diseases with more than 4 million relationships across biological scales.
BioSNAP: Diverse biomedical networks including protein-protein interactions, single-cell similarity networks, and drug-drug interactions.
Genome-wide Perturb-seq: Large-scale perturbation screens (K562 and RPE1 cell lines) enabling systematic study of gene function.
Sciplex3: Single-cell RNA-seq data for over 100 perturbations across multiple cell lines, with dose and time resolution - particularly valuable for training models on chemical perturbations.
Tahoe-100M: A massive dataset of 105 million single-cells across 60,000 conditions for cancer cell lines.
Philosophical Reflections
Beyond the technical talks, ICLR featured several thought-provoking discussions on AI, human intelligence, and psychology. These sessions grappled with fundamental questions about what we’re building and where it’s headed.
One recurring theme was the relationship between artificial and biological intelligence. As our AI systems become more capable, are they converging on similar computational strategies to human cognition, or discovering fundamentally alien approaches to problem-solving? The jury is still out, but the question itself reflects how far the field has come.
Another set of talks focused on AI safety and alignment, particularly “unlearning” techniques for removing harmful capabilities from trained models. As models become more powerful, ensuring they can’t be easily jailbroken to produce harmful outputs becomes increasingly critical. The technical approaches discussed ranged from improved training set filtering to post-hoc unlearning procedures, though no silver bullet has emerged.
Closing Thoughts
ICLR 2025 showcased a field in transition. The move from passive models to active agents, the increasing sophistication of multimodal integration, and the maturation of contrastive learning approaches all point toward AI systems that are more flexible, more powerful, and more practically useful than ever before.
Yet the conference also highlighted persistent challenges: the data quality bottleneck, the difficulty of proper evaluation, and the gap between impressive demos and production systems. The most successful applications will likely come from groups that can address both the algorithmic and the data generation sides of the equation.
For computational biologists and drug discovery researchers, the message is clear: sophisticated AI tools are becoming increasingly accessible (you can run TxGemma on a single GPU!), but generating the right data to train and validate these tools remains the critical challenge. The next AlphaFold won’t come from a better architecture alone - it will require the next PDB.
Singapore provided an awesome setting for the conference: The city’s blend of natural beauty, cutting-edge architecture, and vibrant culture somehow felt fitting for a conference looking toward the future of technology. Between sessions, I could explore hawker centers with incredible Asian cuisine, walk through tropical gardens, or simply marvel at the engineering achievement that is Marina Bay Sands.

The field of deep learning continues to move at a breathtaking pace. If ICLR 2025 is any indication, the next few years should bring AI agents that can meaningfully accelerate scientific discovery - but only if we can match our computational architecture with new sophisticated experimental approaches to generating high-quality data at scale.
Looking forward to my next conference already - maybe ICLR 2026!