Slamdunk paper
For the past couple of years I was involved in the development of SLAMseq, a sequencing technology for time-resolved measurement of newly synthesized and existing RNA in cultured cells. Originally developed by the lab of Stefan Ameres, the lab of my boss Johannes Zuber extended the approach with pharmacological and chemical-genetic perturbations in order to identify direct transcriptional targets of any gene or pathway (Muhar et al, Science 2018).
Processing and interpreting this data required novel analysis methods, so I was given the opportunity to team up with a good friend of mine - Philipp Rescheneder - to develop Slamdunk which we recently published in BMC Bioinformatics and is generally applicable to any nucleotide-conversion containing dataset.
This post will quickly highlight the main functionality, findings and features.
Slamdunk workflow
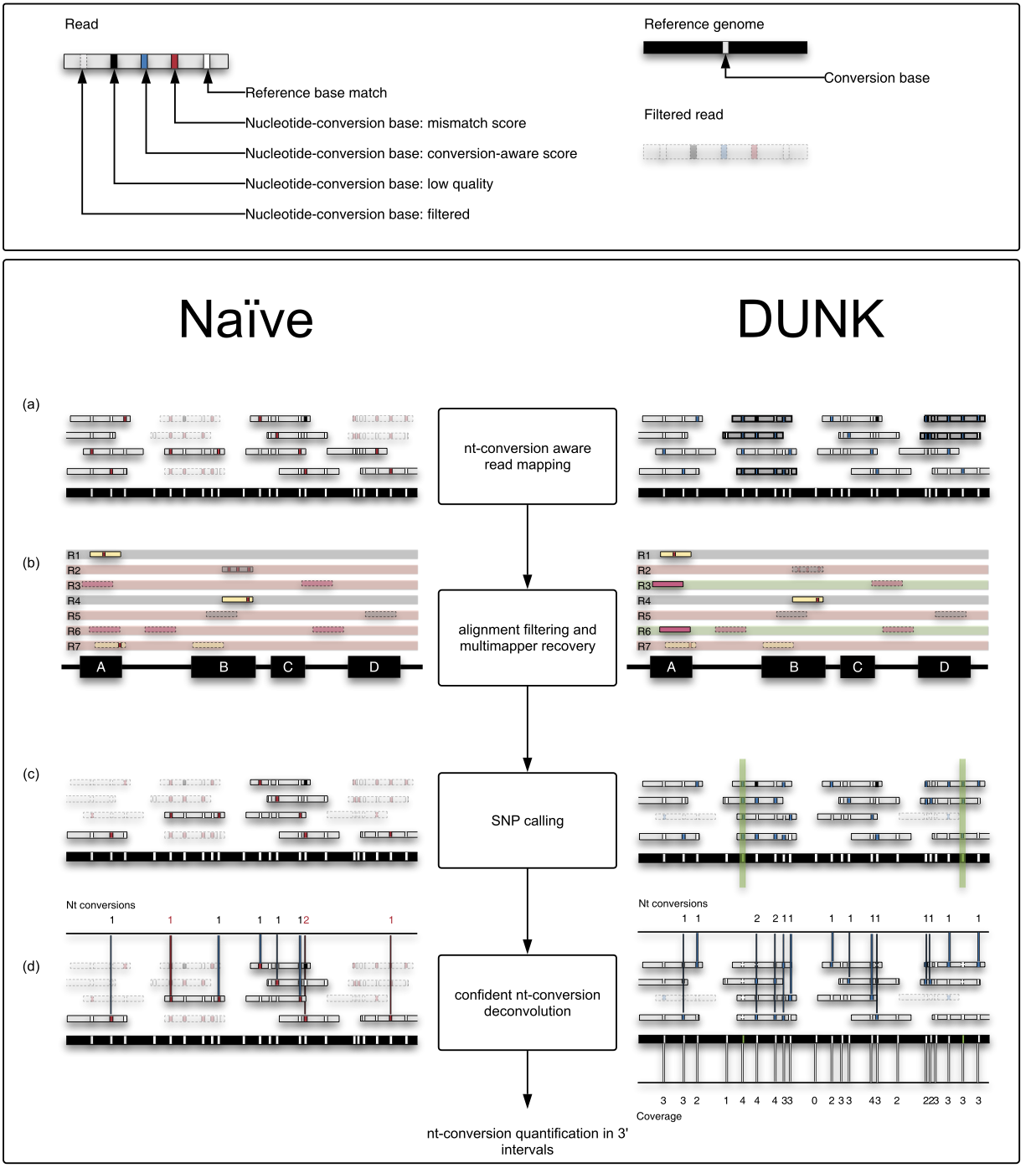
Slamdunk differs from naive read processing in 4 ways:
- It maps with a nucleotide-conversion aware scoring scheme since in the example of SLAMseq data, T>C mismatches are expected and identify reads from labelled transcripts
- Since QuantSeq processes smaller, more repetitive regions of transcripts - namely the 3’ ends - Slamdunk cannot simply discard all multimappers, but utilizes a strategy to recover them
- Genuine T>C SNPs would contribute greatly to false-positive conversion-quantifications and have to be excluded during the quantification step
- Depending on coverage and T-content in the 3’ end, observing T>C reads will have a different likelihood which has to be corrected for during conversion quantification
Features
Conversion-aware mapping
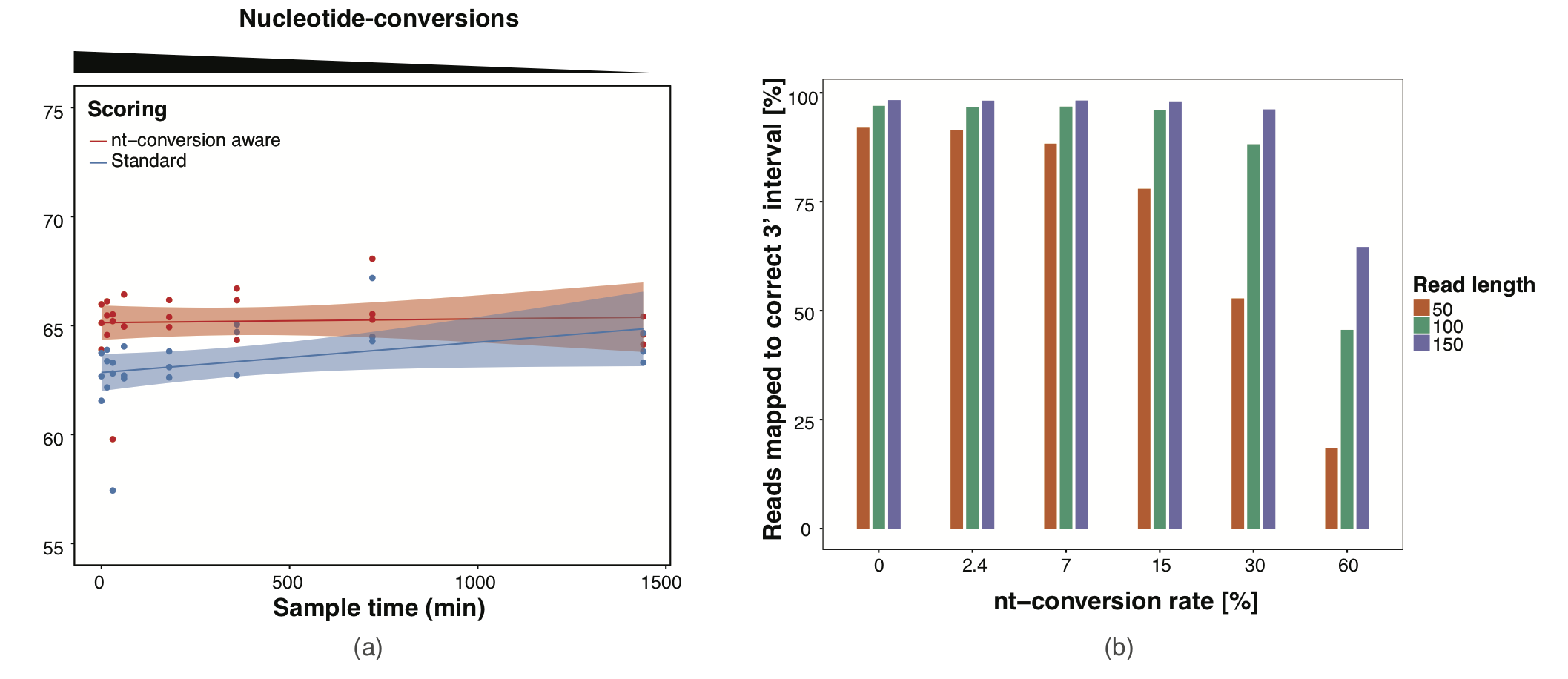
Slamdunk utilizes a conversion-aware scoring scheme implemented with the mapper NextGenMap. Using this scoring-scheme, we could demonstrated the following:
- We can map reads independent of the inherent conversion-rates in the respective datasets (see top Figure a)
- With commonly found conversion-rates (0-7%), we are able to map constantly > 90% of the reads with 100-150bp and >80% of shorter reads with 50bp read length.
Multimapper recovery
We devised a multimapper recovery strategy to deal with repetitive 3’ UTR regions of transcripts. To this end, multimapping reads that still map uniquely to annotated 3’ UTRs are recovered and only reads with alignments to several annotated 3’ UTRs are discarded.
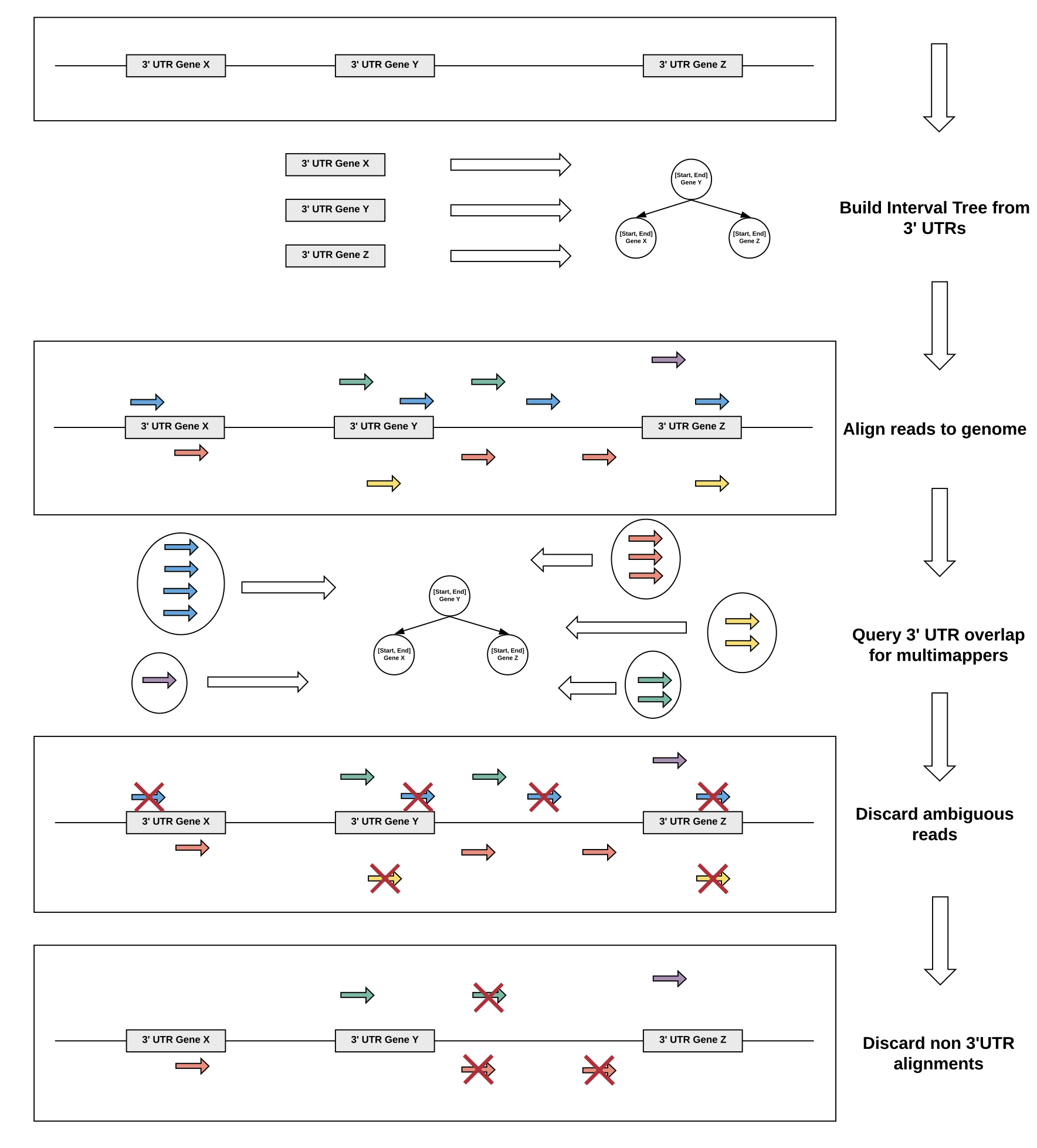
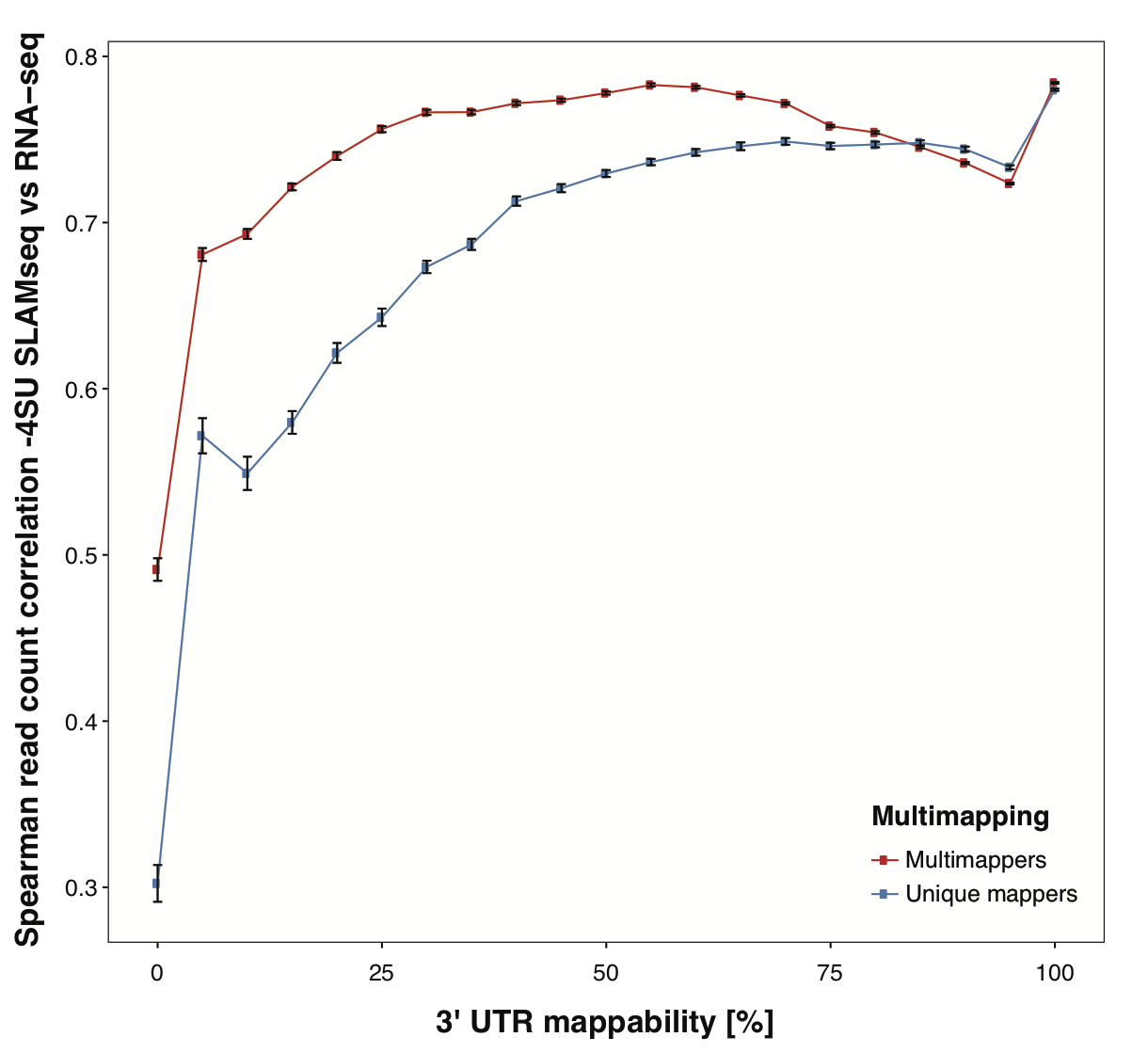
Using this strategy, we are able to recover valuable signal in genes with 3’ UTRs with low mappability and increase overall correlation of QuantSeq datasets to corresponding RNA-seq datasets.
Conversion quantification
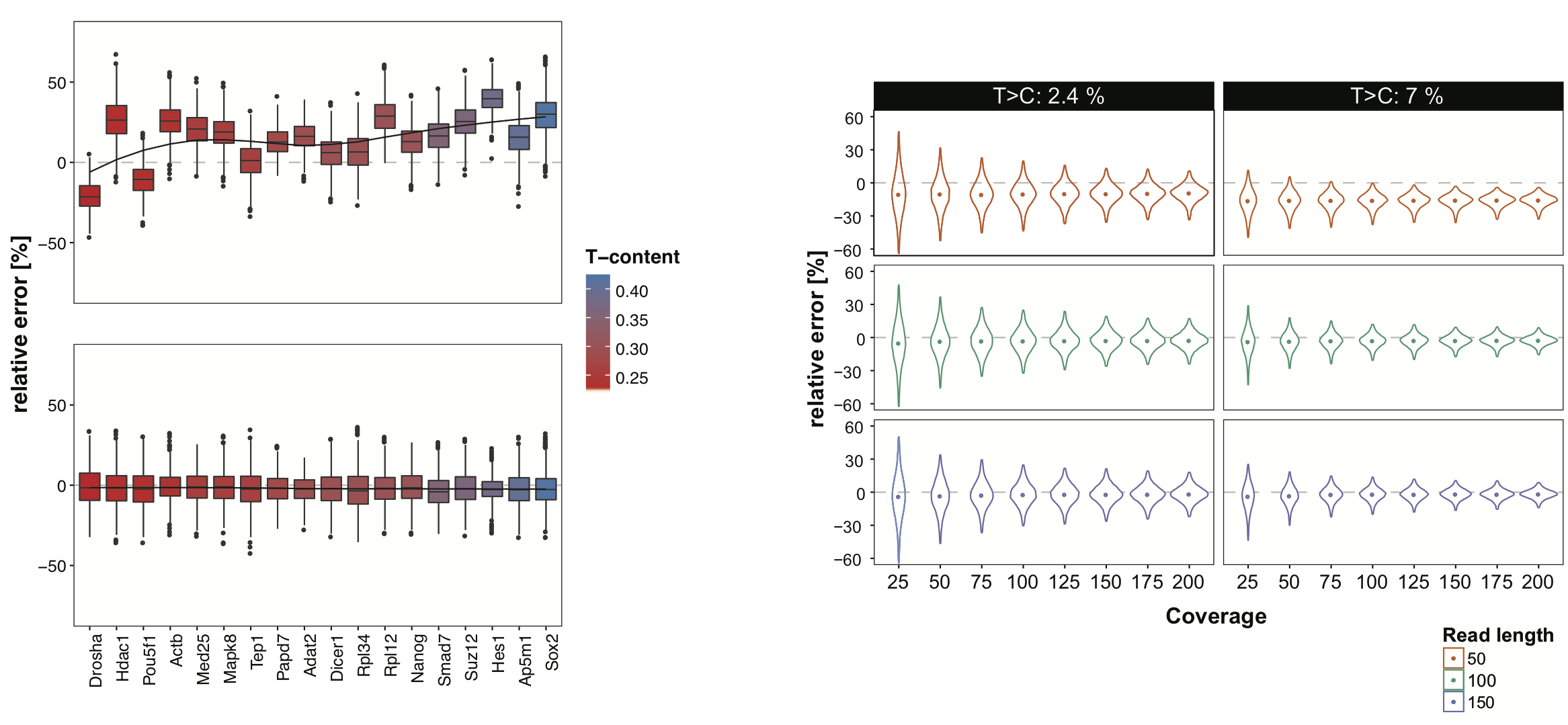
Plain quantification of the number of TC-conversion containing reads in a given interval is biased towards intervals with higher T-content and higher coverage, since the probability of observing a T>C conversion in this intervals is increased. To address this issue, we devised a T-content and coverage aware nucleotide-conversion quantification within intervals that is clearly superior in error rates (see bottom Figure left). Overall, the variance of relative error decreases with higher coverage and while it slightly underestimates the true conversion rate with short reads (50bp), it accurately estimates the conversion rates for reads starting from 100bp (bottom Figure right).
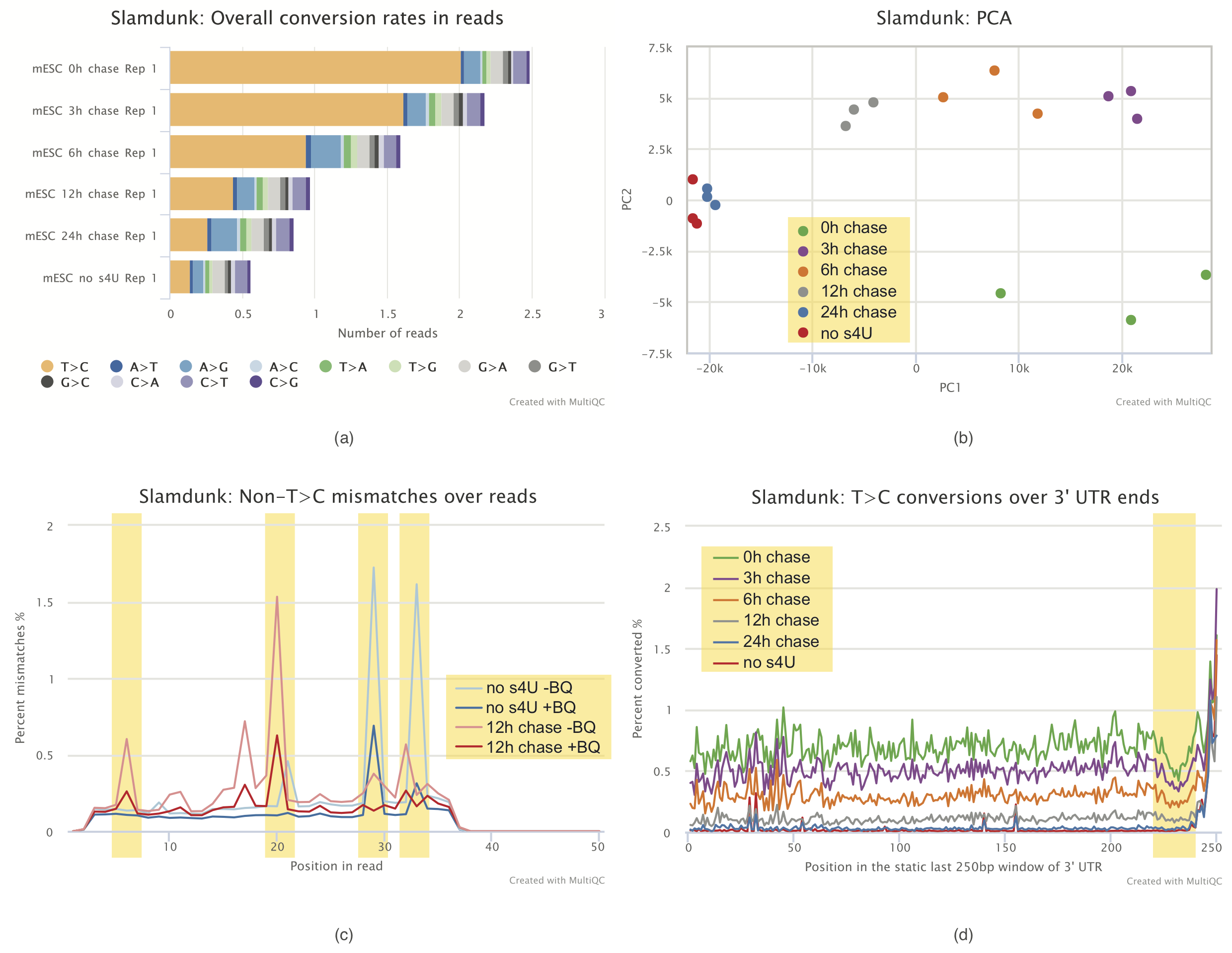
MultiQC report
Visualization of results and quality control is an important aspect of each analysis. To this end, with lots of help from Phil Ewels, we developed a plugin to MultiQC to facilitate quality control of SLAMseq datasets. Using this plugin, we can visualize conversion rates within samples (bottom Figure a), display the principal components of samples based on T>C containing reads (bottom Figure b), plot non T>C mismatches over read positions to identify problematic read positions (bottom Figure c) or plot T>C conversions at 3’ ends (bottom Figure d) to check for base composition biases.
Documentation
A thorough documentation is available from the main website:
Availability
Slamdunk is available from several platforms: