Pipelines with Nextflow
Nowadays, workflow management systems have become an integral part of large-scale analysis of biological datasets with multiple software packages and multi-platform language support. These systems enable the rapid prototyping and deployment of pipelines that combine complementary software packages. Several such systems are already available, such as Snakemake and CWL.
This post will give you an overview of my favourite workflow building system - Nextflow - and look at one toy workflow implementation example that will also be used in later posts.
Nextflow
Here, I will more or less shamelessly copy large parts of the description of Nextflow’s website since it summarises the main features quite neatly.
Up front, the most severe disadvantage for me: Nextflow is written in Groovy which is kind of a pain for me, since I am mostly Python, R, C/C++ and Java based, but have never needed to touch any Groovy.
However, with some fiddling around and especially a lot of low-latency community support via the Nextflow Gitter channel, these are hurdles that can be overcome.
Once you lost your fear of Groovy, the advantages of Nextflow are quite convincing.
If you want to read more about Nextflow, here is the documentation and here is the original paper.
Fast prototyping
Nextflow allows you to write a computational pipeline by making it simpler to put together many different tasks.
You may reuse your existing scripts and tools and you don’t need to learn a new language or API to start using it.
As an example, look at how easy it is to run code from different languages within Nextflow processes out of the box.
process perlStuff {
"""
#!/usr/bin/perl
print 'Hi there!' . '\n';
"""
}
process pyStuff {
"""
#!/usr/bin/python
x = 'Hello'
y = 'world!'
print "%s - %s" % (x,y)
"""
}
Portable
Nextflow provides an abstraction layer between your pipeline’s logic and the execution layer, so that it can be executed on multiple platforms without it changing.
It provides out of the box executors for SGE, LSF, SLURM, PBS and HTCondor batch schedulers and for Kubernetes, Amazon AWS and Google Cloud platforms.
Again, check the so-called profile configurations one can quite easily setup to enable support for yet another scheduler.
profiles {
standard {
process.executor = 'local'
}
cluster_sge {
process.executor = 'sge'
process.penv = 'smp'
process.cpus = 20
process.queue = 'public.q'
process.memory = '10GB'
}
cluster_slurm {
process.executor = 'slurm'
process.cpus = 20
process.queue = 'work'
}
}
With these few lines of code, you can now seamlessly execute your pipeline on your local machine, on PBS and SLURM, even with customized resource settings.
Reproducibility
Nextflow supports Docker and Singularity containers technology.
This, along with the integration of the GitHub code sharing platform, allows you to write self-contained pipelines, manage versions and to rapidly reproduce any former configuration.
This is an especially nice feature, since it also allows to run Nextflow workflows on cloud based platforms such as Amazon Web Services which strictly require all software environments supplied in a public Docker registry reachable by ECS batch.
Unified parallelism
Nextflow is based on the dataflow programming model which greatly simplifies writing complex distributed pipelines.
Parallelisation is implicitly defined by the processes input and output declarations. The resulting applications are inherently parallel and can scale-up or scale-out, transparently, without having to adapt to a specific platform architecture.
Continuous checkpoints
All the intermediate results produced during the pipeline execution are automatically tracked.
This allows you to resume its execution, from the last successfully executed step, no matter what the reason was for it stopping.
Stream oriented
Nextflow extends the Unix pipes model with a fluent DSL, allowing you to handle complex stream interactions easily.
It promotes a programming approach, based on functional composition, that results in resilient and easily reproducible pipelines.
Salmon
Our first small toy Nextflow workflow will be based upon Salmon.
Salmon is a tool for quantifying the expression of transcripts using RNA-seq data. Salmon uses the concept of quasi-mapping coupled with a two-phase inference procedure to provide accurate expression estimates very quickly (i.e. wicked-fast) and while using little memory. Salmon performs its inference using an expressive and realistic model of RNA-seq data that takes into account experimental attributes and biases commonly observed in real RNA-seq data.
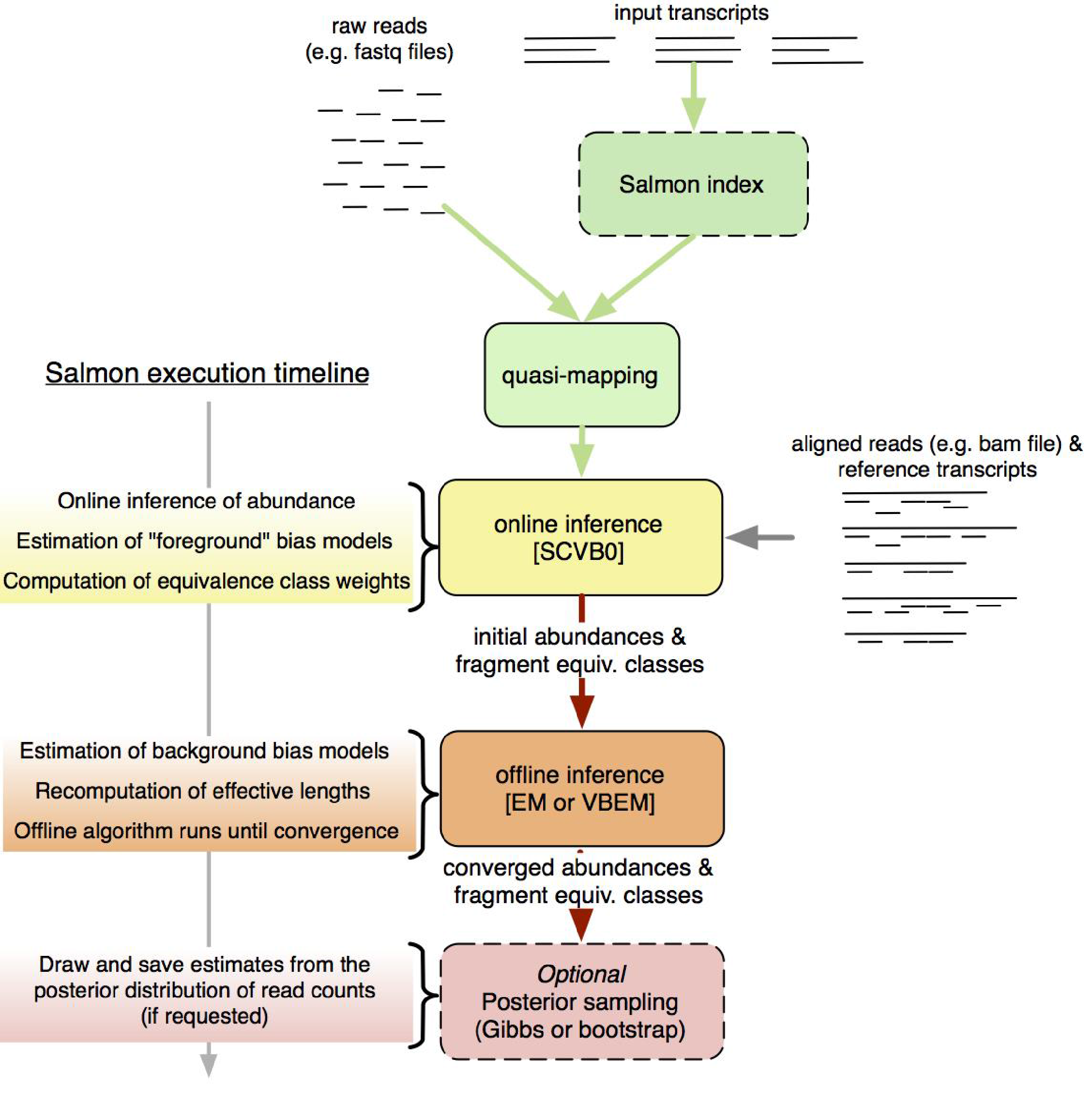
Essentially, Salmon will create a transcript index which it then uses to quantify expression estimates for each of the transcripts from raw fastq reads.
Our goal:
- Obtain those transcript expression estimates for our samples
- Obtain reads mapping to these transcripts via the
--writeMappingsflag as pseudo-bam
If you want to read more on Salmon, here is the paper.
salmon-nf
So the Nextflow pipeline we will create during this exercise I will call salmon-nf and it can be found on my GitHub page as a fully functional repository.
Any standalone Nextflow pipeline will need 2 files to be executable out of the box and also directly from GitHub:
main.nf- This file contains the individual processes and channelsnextflow.config- The configuration file for parameters, profiles etc. For more info read here
Workflow layout
First, we need to get an idea about what the data flow will be and what software and scripts will be run on it. I have outline the basic workflow of salmon-nf below:
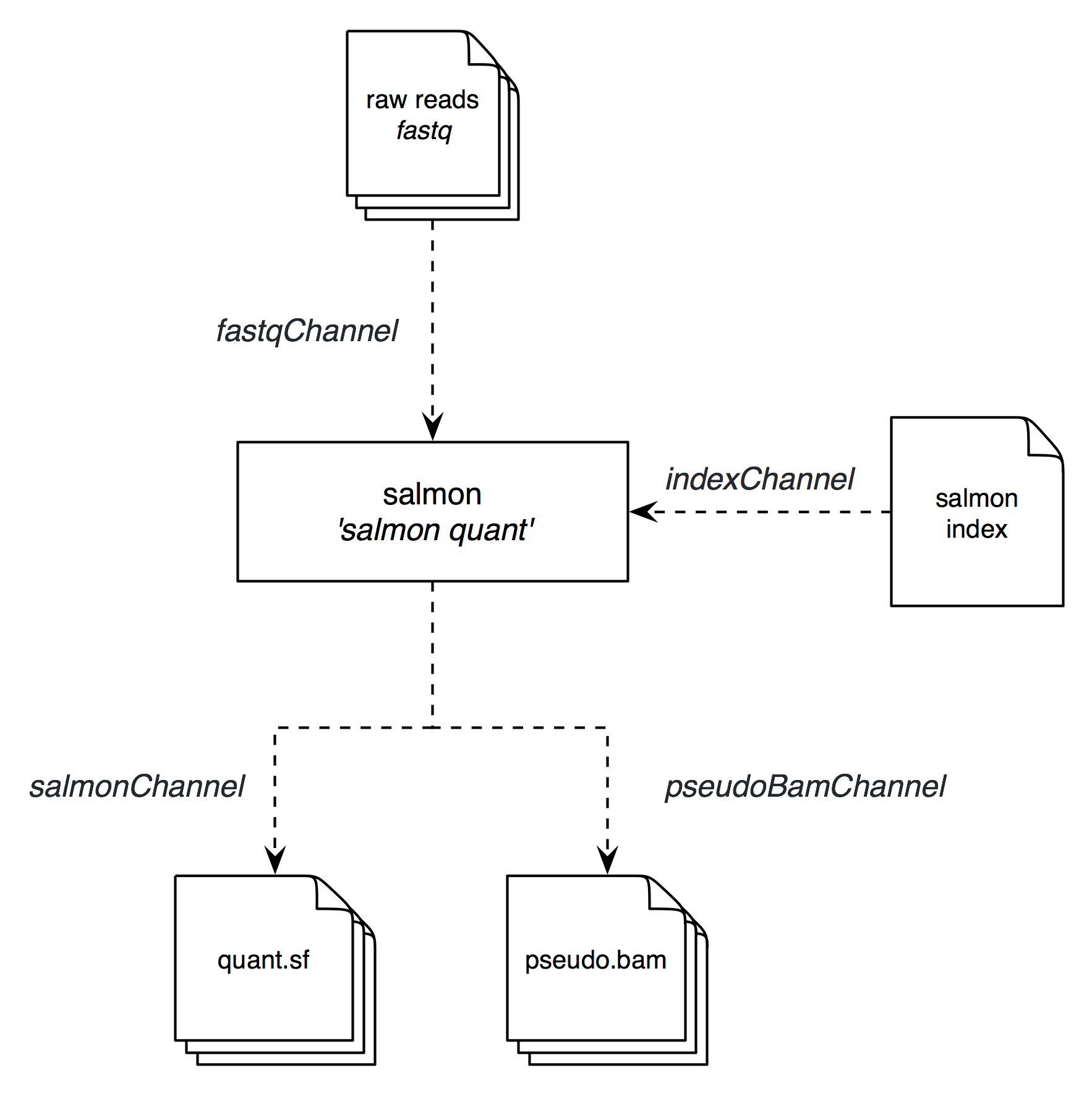
We will only have one single process salmon which will use the input fastq files and the respective transcriptome index file to produce our expression estimates and the pseudo-bam files of aligning reads.
So for our salmon process we will have 2 input channels:
fastqChannel- feeding in our raw reads infastqformatindexChannel- providing our transcriptomeindexto which we align the reads to
Our salmon process will produce several output files of which we choose to feed 2 file types into output processes as our final results:
quant.sffiles via thesalmonChanneloutput channelpseudo.bamfiles via thepseudoBamChanneloutput channel
Now let’s have a look how we can actually realize and implement this on the coding end.
Docker container
Before we can run anything, we need to provide the software environment containing all dependencies and software packages our salmon process will run. These days, this is usually done via a Docker container, or a Singularity container on HPC environments.
Many software packages - including Salmon in our case - usually provide already read-to-use Docker containers (combinelab/salmon). But even if they don’t, do not despair and brainlessly jump into creating your own containers. If the packages was provided via BioConda, you will find a Docker container on BioContainers. I found this last resort to work in many cases.
Either way, since I wanted to convert the raw SAM output from salmon into a compressed BAM file, I chose to extend their Docker image with adding samtools as shown in the Dockerfile below.
# Copyright (c) 2019 Tobias Neumann.
#
# You should have received a copy of the GNU Affero General Public License
# along with this program. If not, see <http://www.gnu.org/licenses/>.
FROM combinelab/salmon:0.12.0
MAINTAINER Tobias Neumann <tobias.neumann.at@gmail.com>
RUN buildDeps='wget ca-certificates make g++' \
runDeps='zlib1g-dev libncurses5-dev unzip gcc' \
&& set -x \
&& apt-get install -y $buildDeps $runDeps --no-install-recommends \
&& rm -rf /var/lib/apt/lists/* \
&& wget https://github.com/samtools/samtools/releases/download/1.9/samtools-1.9.tar.bz2 \
&& tar xvfj samtools-1.9.tar.bz2 \
&& cd samtools-1.9 \
&& ./configure --prefix=/usr/local/ \
&& make \
&& make install \
&& apt-get purge -y --auto-remove $buildDeps
The resulting Docker image was pushed to Docker Hub and can be pulled via docker pull obenauflab/salmon:latest.
main.nf
Now we are ready to create the central main.nf file which contains all processes as well as channels. As mentioned before, you will find the entire code on GitHub, so here is an excerpt of the important sections.
fastqChannel
pairedEndRegex = params.inputDir + "/*_{1,2}.fq.gz"
SERegex = params.inputDir + "/*[!12].fq.gz"
pairFiles = Channel.fromFilePairs(pairedEndRegex)
singleFiles = Channel.fromFilePairs(SERegex, size: 1){ file -> file.baseName.replaceAll(/.fq/,"") }
singleFiles.mix(pairFiles)
.set { fastqChannel }
This elaborate chunk of code is needed to enable the fastqChannel input channel to our salmon process to handle both single- and paired-end fastq files. As you can see, we created a pairFiles channel with a paired-end regex basically assuming that our read-pairs are named *_1.fq.gz and *_2.fq.gz. In addition, we have a singleFiles channel that takes all fastq files not following the _1 and _2 naming convention and assuming it is single-end read files.
The fromFilePairs method creates a channel emitting the file pairs matching the regex we provided. The matching files are emitted as tuples in which the first element is the grouping key of the matching pair and the second element is the list of files (sorted in lexicographical order). For example:
[0399ad16-816f-4824-ae28-7b82e006e7b7_gdc_realn_rehead, [0399ad16-816f-4824-ae28-7b82e006e7b7_gdc_realn_rehead_1.fq.gz, 0399ad16-816f-4824-ae28-7b82e006e7b7_gdc_realn_rehead_2.fq.gz]]
[0ac6634e-00b0-4107-a5d6-db8ffc602645_gdc_realn_rehead, [0ac6634e-00b0-4107-a5d6-db8ffc602645_gdc_realn_rehead_1.fq.gz, 0ac6634e-00b0-4107-a5d6-db8ffc602645_gdc_realn_rehead_2.fq.gz]]
As you can see, for the single-end reads channel singleFiles, the method is slightly extended:
First, we set an additional parameter size: 1 to set the number of files each emitted item is expected to hold to 1. In additional, we manually provide the a custom grouping strategy in the closure, which based on the current file as parameter, returns the grouping key. In our case, we simply strip anything from the file name after .fq and use this as our grouping key. For example:
[0fdb3d0e-e405-4e8d-8897-4a90ea4fe00c_gdc_realn_rehead, [0fdb3d0e-e405-4e8d-8897-4a90ea4fe00c_gdc_realn_rehead.fq.gz]]
[1916abcd-61c0-4f23-96ac-be70aacb8dc1_gdc_realn_rehead, [1916abcd-61c0-4f23-96ac-be70aacb8dc1_gdc_realn_rehead.fq.gz]]
Finally, we combined both channels via a mix operator into our final fastqChannel input channel to our salmon process.
indexChannel
indexChannel = Channel
.fromPath(params.salmonIndex)
.ifEmpty { exit 1, "Salmon index not found: ${params.salmonIndex}" }
This input channel is pretty straightforward set up. Only thing we need to do is to precreate our Salmon index (read how to do this here) and supply it via the salmonIndex parameter - how this is done will follow later.
Process salmon
process salmon {
tag { lane }
input:
set val(lane), file(reads) from fastqChannel
file index from indexChannel.first()
output:
file ("${lane}_salmon/quant.sf") into salmonChannel
file ("${lane}_pseudo.bam") into pseudoBamChannel
shell:
def single = reads instanceof Path
if (!single)
'''
salmon quant -i !{index} -l A -1 !{reads[0]} -2 !{reads[1]} -o !{lane}_salmon -p !{task.cpus} --validateMappings --no-version-check -z | samtools view -Sb -F 256 - > !{lane}_pseudo.bam
'''
else
'''
salmon quant -i !{index} -l A -r !{reads} -o !{lane}_salmon -p !{task.cpus} --validateMappings --no-version-check -z | samtools view -Sb -F 256 - > !{lane}_pseudo.bam
'''
}
Our only process for the salmon-nf workflow is the salmon process.
You will notice that it has the 2 input channels we previously defined - fastqChannel and indexChannel. Note, how we have to use the .first() method on the indexChannel since it is a folder.
In addition, we have defined 2 output channels - salmonChannel outputting all quant.sf files and pseudoBamChannel outputting the corresponding pseudo.bam files.
The actual script that is run, is a plain conditional bash script. We have an initial condition that asks whether we have single read files coming in from the fastqChannel or paired-end reads - and based on this evaluation will run one or the other script branch.
The bash script itself is then basically only a salmon call on the respective input files.
This input channel is pretty straightforward set up. Only thing we need to do is to precreate our Salmon index (read how to do this here) and supply it via the salmonIndex parameter - how this is done will follow later.
nextflow.config
The Nextflow configuration files contain directives for for parameter definitions, profile definitions and many others.
In our particular example of salmon-nf, we will have a master nextflow.config that is tidied up and include additional configs for each section.
includeConfig 'config/general.config'
includeConfig 'config/docker.config'
profiles {
standard {
process.executor = 'local'
process.maxForks = 3
}
slurm {
includeConfig 'config/slurm.config'
}
awsbatch {
includeConfig 'config/awsbatch.config'
}
}
As you can see, we have simply included some more config files and some barebone definition of profiles. Let’s look at the sub-config files.
general.config
This holds general configurations, parameters and definitions that are applicable to any of our run profiles.
params {
outputDir = './results'
}
process {
publishDir = [
[path: params.outputDir, mode: 'copy', overwrite: 'true', pattern: "*/quant.sf"],
[path: params.outputDir, mode: 'copy', overwrite: 'true', pattern: "*pseudo.bam"]
]
errorStrategy = 'retry'
maxRetries = 3
maxForks = 100
}
cloud {
imageId = 'ami-0f99d00928be3a282'
instanceType = 't2.micro'
userName = 'ec2-user'
keyName = 'awsbatch'
// Type: SSH, Protocol: TCP, Port: 22, Source IP: 0.0.0.0/0
securityGroup = 'sg-0307dbec406526c14'
}
timeline {
enabled = true
}
report {
enabled = true
}
We set a default output directory in the params section, copy the quant.sf and pseudo.bam files to a dedicated publish directory, set our error strategy, a basic cloud profile for starting up instances on AWS and enable timeline and execution reports per default.
docker.config
With this configuration file, we enable Docker support per default and supply the Docker image to use with our salmon process.
docker {
enabled = true
}
process {
// Process-specific docker containers
withName:salmon {
container = 'obenauflab/salmon:latest'
}
}
slurm.config
This configuration file defines a profile for the SLURM scheduler which is run on our HPC system. Our cluster only supports Singularity, so we disable Docker and enable Singuarity in return, as well as define basic resource constraints and queues on our HPC system where to run our tasks - and finally also supply the location of the salmonIndex on our file system.
singularity {
enabled = true
}
docker {
enabled = false
}
process {
executor = 'slurm'
clusterOptions = '--qos=short'
cpus = '12'
memory = { 8.GB * task.attempt }
}
params {
salmonIndex = '/groups/Software/indices/hg38/salmon/gencode.v28.IMPACT'
}
awsbatch.config
This configuration file will be explained in detail in a later post - but in brief it enables execution of tasks in the cloud using AWS Batch, yet it still requires extensive configuration before it is usable.
Running the salmon-nf Nextflow workflow
Now that we have written our code and committed everything to GitHub, we can finally testdrive our workflow on some actual data.
First, let’s pull in our workflow:
tobias.neumann@login-01 [BIO] $ nextflow pull t-neumann/salmon-nf
Picked up _JAVA_OPTIONS: -Djava.io.tmpdir=/tmp
Checking t-neumann/salmon-nf ...
downloaded from https://github.com/t-neumann/salmon-nf.git - revision: 4fbaea7165 [master]
tobias.neumann@login-01 [BIO] $
Now we are ready to run our workflow. Make sure to select the profile you desire - for this example I will run it on our in-house cluster with SLURM:
tobias.neumann@login-01 [BIO] $ nextflow run t-neumann/salmon-nf --inputDir /tmp/data --outputDir results -profile slurm -resume
Picked up _JAVA_OPTIONS: -Djava.io.tmpdir=/tmp
N E X T F L O W ~ version 19.01.0
Launching `t-neumann/salmon-nf` [maniac_poisson] - revision: 4fbaea7165 [master]
parameters
======================
input directory : /tmp/data
output directory : results
======================
[warm up] executor > slurm
[fb/20d1dc] Submitted process > salmon (8cec7235-3572-460c-b1d7-efe7961988e1_gdc_realn_rehead)
[e9/6f6404] Submitted process > salmon (5e18b02d-7e56-4f0d-b892-e7798eee5205_gdc_realn_rehead)
[f9/509312] Submitted process > salmon (d1ada222-b67f-47c0-b380-091eaab093b4_gdc_realn_rehead)
[6d/30354f] Submitted process > salmon (3783843f-c4fa-4aab-8f5b-e0749763164e_gdc_realn_rehead)
[9b/2a81e9] Submitted process > salmon (0fdb3d0e-e405-4e8d-8897-4a90ea4fe00c_gdc_realn_rehead)
[de/418130] Submitted process > salmon (383e3574-d22c-4dd6-842f-656ee2ab3b32_gdc_realn_rehead)
[c1/e00c04] Submitted process > salmon (1916abcd-61c0-4f23-96ac-be70aacb8dc1_gdc_realn_rehead)
[63/6a2e93] Submitted process > salmon (30fe4005-f4f2-41ce-bb1a-4830f3959ab7_gdc_realn_rehead)
Now we just have to wait till our workflow has successfully finished processing all our samples.
[76/67754e] Submitted process > salmon (0399ad16-816f-4824-ae28-7b82e006e7b7_gdc_realn_rehead)
t-neumann/salmon-nf has finished.
Status: SUCCESS
Time: Sun Aug 25 23:35:49 CEST 2019
Duration: 2m
tobias.neumann@login-01 [BIO] $
If we now check our results and execution folder, we will find all the files we asked for in there - Nextflow is awesome!
tobias.neumann@login-01 [BIO] $ ls
report.html results timeline.html
tobias.neumann@login-01 [BIO] $ ls results
0399ad16-816f-4824-ae28-7b82e006e7b7_gdc_realn_rehead_pseudo.bam 0399ad16-816f-4824-ae28-7b82e006e7b7_gdc_realn_rehead_salmon
Have fun building workflows on your own - it pays off, especially for larger samples and heterogeneous computing environments!